Clear Sky Science · sv
DIS3‑mutationer ökar AID‑drivna translokationer under B‑cellsaktivering och främjar omvandling till multipelt myelom
När väsentlig cellstädning går fel
Vårt immunsystem är beroende av B‑celler för att tillverka antikroppar som bekämpar infektioner. För att göra detta skär B‑celler medvetet i sitt eget DNA och syr sedan ihop det igen — en riskfylld process som ibland kan gå snett och leda till blodcancer såsom multipelt myelom. Denna studie avslöjar hur subtila förändringar i ett centralt "RNA‑städnings"‑enzym kallat DIS3 tyst kan rubba denna balans mot cancer, genom att öka farliga DNA‑omläggningar utan att tydligt störa normal immunfunktion.
En tyst mutation med dolda konsekvenser
DIS3‑enzymet hjälper celler att göra sig av med kortlivade RNA‑fragment i kärnan, bland annat de som bildas när B‑celler omformar sina antikroppsgen. Hos patienter med multipelt myelom bär 10–20 % av dem mutationer som försvagar DIS3:s förmåga att bryta ner RNA, men det var oklart hur detta driver cancer. Forskarna fokuserade på en kliniskt observerad DIS3‑ändring, kallad G766R, och byggde en musmodell där endast en av de två Dis3‑kopiorna bar denna variant. Dessa möss såg friska ut: deras blodceller utvecklades normalt, gen‑ och proteingradienten var bara subtilt förändrade, och nyckelprocesser i immunförsvaret såsom klassbyte och antikroppsfinkalibrering fungerade som vanligt.
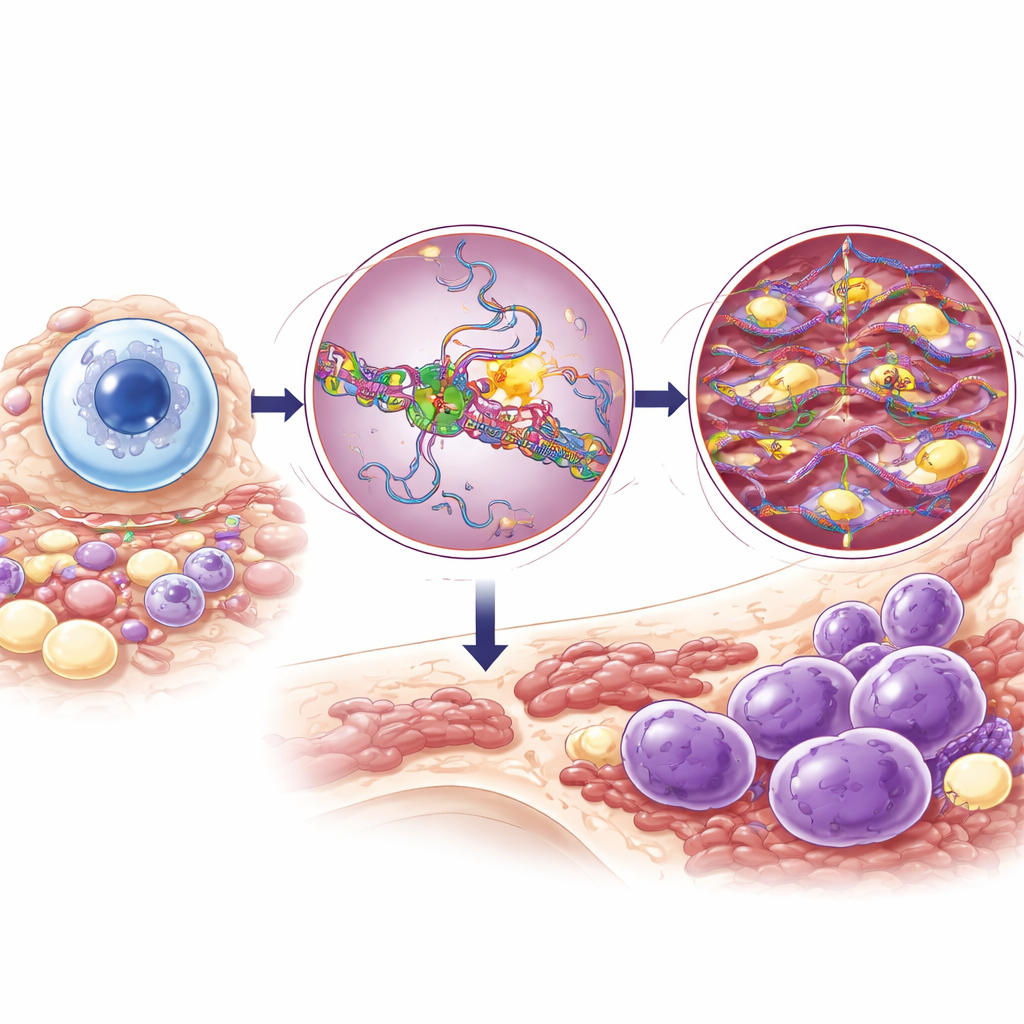
DNA‑redigering i B‑celler och DIS3:s roll
När B‑celler aktiveras skadar ett enzym kallat AID med flit DNA i antikroppsgener så att cellen kan byta antikropstyp och förbättra deras precision. För att göra detta säkert måste AID vara hårt kontrollerat och huvudsakligen begränsat till antikroppslokuset. DIS3 hjälper normalt till genom att avlägsna nyligen syntetiserat RNA nära aktiva gener, vilket blottar rätt DNA‑sträng så att AID kan verka fokuserat och temporärt. Om AID vandrar till andra delar av genomet kan det orsaka kromosombrott och omläggningar som aktiverar cancerfrämjande gener. Tidigare arbete antydde att fullständig förlust av DIS3 slår sönder detta system och blockerar normalt klassbyte; detta väckte frågan varför patienter bär partiella DIS3‑mutationer istället för total förlust.
Mutant DIS3 ökar risken för kromosomväxlingar
För att undersöka cancerrisken exponerade teamet Dis3G766R/+‑möss för pristane, en kemikalie som främjar kronisk inflammation och plasmacytom, en mustumör lik tidigt multipelt myelom. På en genetisk bakgrund som normalt är motståndskraftig mot denna sjukdom utvecklade möss med mutant DIS3 plasmacytom mycket oftare och snabbare än normala möss. Djup DNA‑sekvensering av dessa tumörer visade en kraftig ökning av kromosomtranslokationer som involverade antikropparnas tunga kedjeregion (IGH), ett kännetecken för mänskligt multipelt myelom. Liknande mönster framträdde när författarna analyserade stora patientdatamängder: individer med DIS3‑mutationer hade mycket större sannolikhet att bära IGH‑translokationer och att visa AID‑liknande mutationssignaturer i kända myelom‑driver‑gener, även om deras övergripande mutationsmönster över genomet liknade patienter utan DIS3‑mutationer.
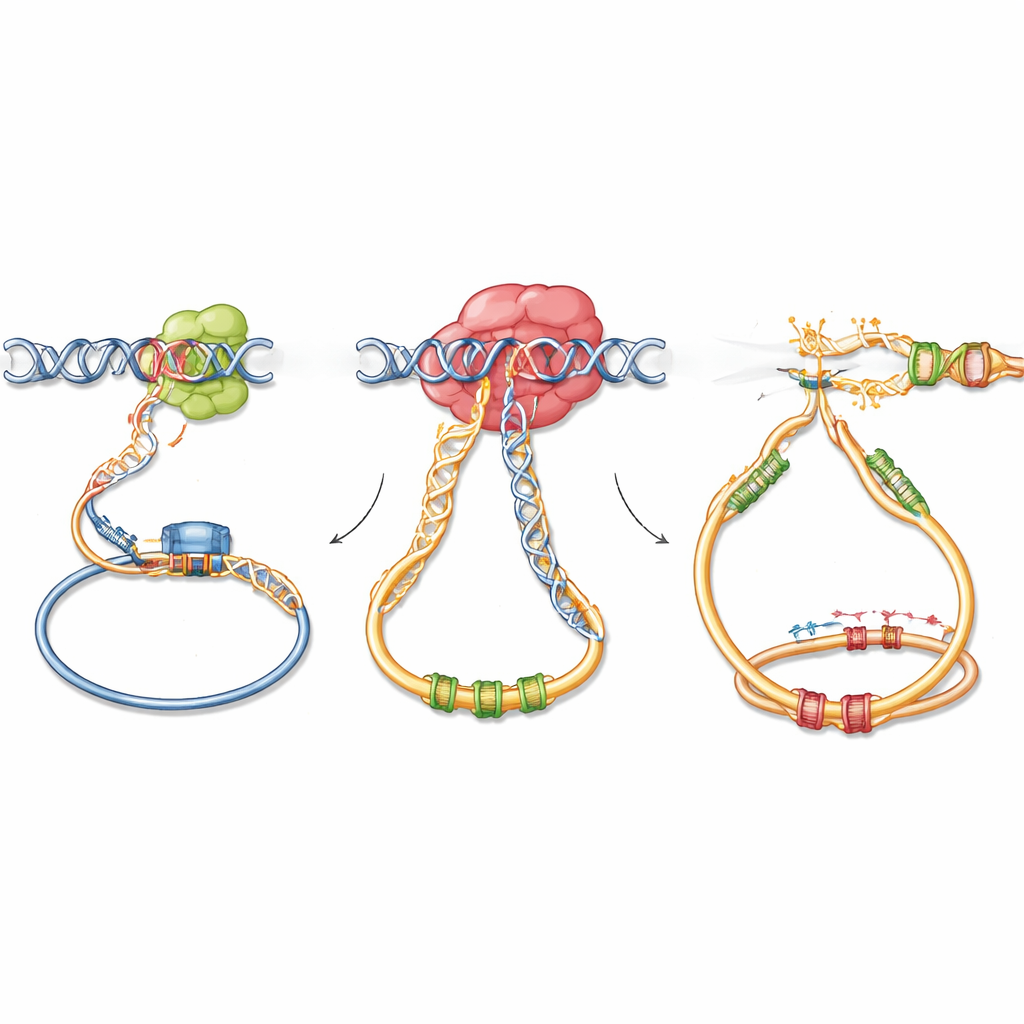
Hur fastklistrad RNA‑städning främjar DNA‑skador
Mechanistiska experiment visade vad som går fel på molekylär nivå. G766R‑varianten av DIS3 fungerar fortfarande på enkla RNA men tenderar att fastna på strukturerat RNA och RNA–DNA‑hybrider nära aktiva gener och regulatoriska regioner. Med en känslig kartläggningsteknik fann författarna att mutant DIS3 ackumuleras starkare på kromatin vid platser där AID också rekryteras, särskilt runt antikropps‑switch‑regioner, enhancer‑element och kända AID‑off‑target‑gener. Vid dessa platser byggs RNA–DNA‑hybrider och ovanliga DNA‑strukturer upp, vilket blottar sträckor av enkelsträngat DNA på båda strängarna. I aktiverade B‑celler och i modellcellinjer ledde detta till fler DNA‑skadefoci, men endast när AID var närvarande. Viktigt är att den vanliga tendensen hos AID att föredra en DNA‑sträng försvann, vilket gav mer symmetrisk skada på båda strängarna — ett läge som gynnar upptrappade dubbelsträngsbrott och felbenägen reparation som syr samman missanpassade DNA‑ändar.
Kapning av normal genomarkitektur
Studien undersökte också om DIS3‑mutationen omformar DNA:s tredimensionella veckning i kärnan, vilket är avgörande för att samla antikroppsgener och deras regulatoriska element. Högupplösta mätningar av kromosomkonformation visade att, till skillnad från fullständig förlust av DIS3, lämnar G766R‑varianten den övergripande arkitekturen i antikroppslokuset intakt. Istället för att omkoppla genomet verkar mutant DIS3 utnyttja befintliga fysiska kontakter: translokationsbrytpunkter i musstumörer klustrar nära aktiva promotorer, enhancers och bindningsställen för arkitektoniska proteiner som definierar kromatinloopar. I praktiken ökar mutationen sannolikheten att när AID‑inducerade brott uppstår vid dessa naturligt närliggande platser, kommer de att återfogas felaktigt och permanent länka starka antikroppsenhancers till närliggande cancerfrämjande gener.
Från subtilt enzymbyte till blodcancer
Sammantaget visar detta arbete att DIS3‑mutationer kopplade till multipelt myelom inte verkar genom att brett störa B‑cellsbiologi, utan genom att skärpa en mycket specifik risk under korta fönster av B‑cellsaktivering. Det mutanta enzymet dröjer kvar på strukturerade RNA vid AID‑mål, ökar AID:s åtkomst till båda DNA‑strängarna och höjer sannolikheten för felreparerade dubbelsträngsbrott som förenar antikroppsregioner med onkogener. Eftersom normalt klassbyte och B‑cellsvar i stort förblir intakta kan dessa "gain‑of‑function"‑DIS3‑varianter bestå tillräckligt länge för att så småningom ge upphov till de kromosomala omläggningar som driver multipelt myelom.
Citering: Kuliński, T.M., Gewartowska, O., Mahé, M. et al. DIS3 mutations enhance AID-driven translocations during B-cell activation, promoting transformation to multiple myeloma. Nat Commun 17, 3976 (2026). https://doi.org/10.1038/s41467-026-70386-3
Nyckelord: multipelt myelom, B‑celler, kromosomala translokationer, RNA‑exosom, aktiveringsinducerad cytidin deaminas