Clear Sky Science · en
DIS3 mutations enhance AID-driven translocations during B-cell activation, promoting transformation to multiple myeloma
When Essential Cell Clean-Up Goes Wrong
Our immune system relies on B cells to produce antibodies that fight infections. To do this, B cells deliberately cut and re‑stitch their own DNA, a risky process that can sometimes go awry and lead to blood cancers such as multiple myeloma. This study uncovers how subtle changes in a key “RNA clean‑up” enzyme called DIS3 can quietly tip this balancing act toward cancer, increasing dangerous DNA rearrangements without obviously harming normal immune function.
A Quiet Mutation with Hidden Consequences
The DIS3 enzyme helps cells dispose of short‑lived RNA fragments in the nucleus, including those produced when B cells reshape their antibody genes. In multiple myeloma, 10–20% of patients carry mutations that weaken DIS3’s ability to chew up RNA, but it was unclear how this drives cancer. The researchers focused on a clinically observed DIS3 change, called G766R, and built a mouse model in which just one of the two Dis3 copies carries this variant. These mice looked healthy: their blood cells developed normally, gene and protein activity were only subtly altered, and key immune processes such as class switching and antibody fine‑tuning worked as usual.
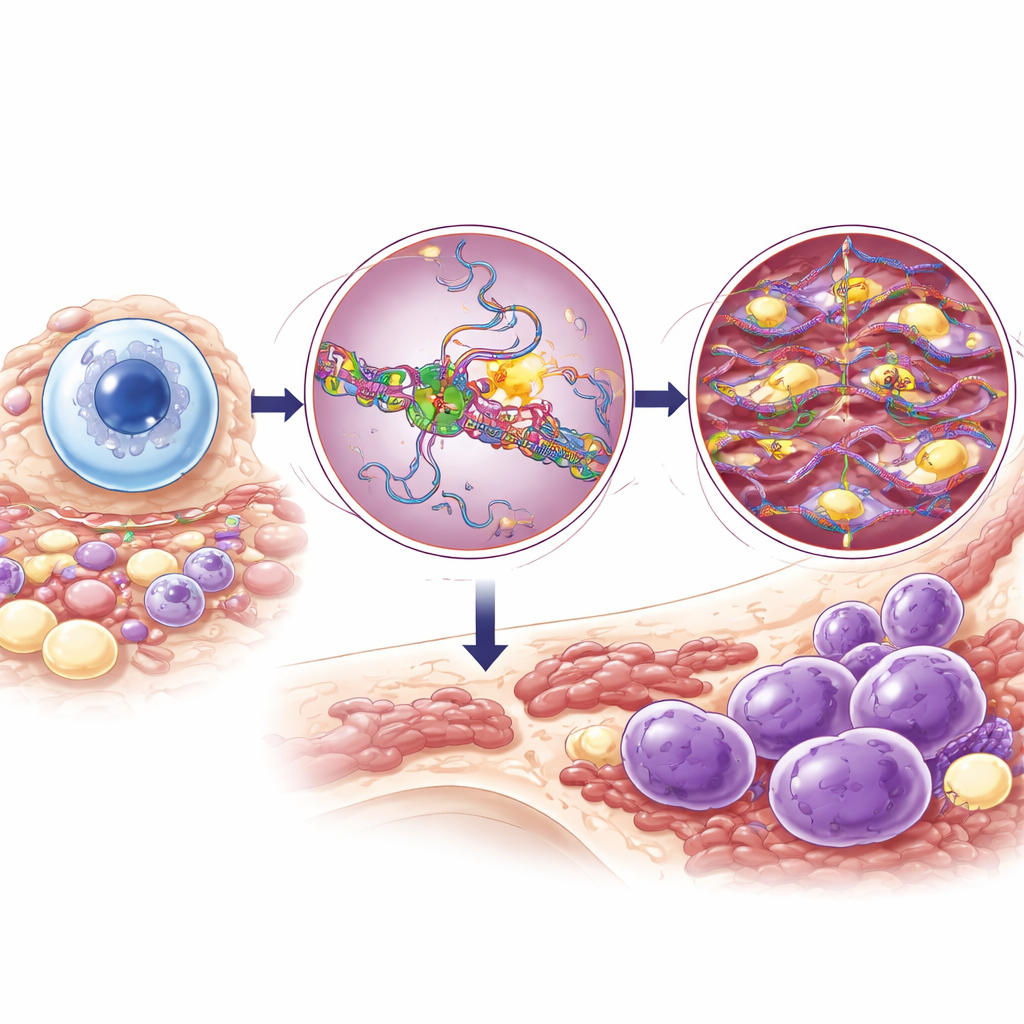
DNA Editing in B Cells and the Role of DIS3
When B cells are activated, an enzyme called AID intentionally damages DNA in antibody genes so the cell can switch antibody types and improve their precision. To do this safely, AID must be tightly controlled and mainly confined to the antibody locus. DIS3 normally helps by removing newly made RNA near active genes, exposing the correct DNA strand so AID can work in a focused and temporary way. If AID strays to other parts of the genome, it can trigger chromosome breaks and rearrangements that activate cancer‑promoting genes. Earlier work suggested that completely eliminating DIS3 wrecks this system and blocks normal antibody switching; this led to the question of why patients carry partial DIS3 mutations instead of total loss.
Mutant DIS3 Raises the Risk of Chromosome Swaps
To probe the cancer risk, the team exposed the Dis3G766R/+ mice to pristane, a chemical that promotes chronic inflammation and plasmacytoma, a mouse tumor similar to early multiple myeloma. On a genetic background usually resistant to this disease, mice with the mutant DIS3 developed plasmacytomas much more often and more rapidly than normal mice. Deep DNA sequencing of these tumors showed a spike in chromosome translocations involving the antibody heavy chain region (IGH), a hallmark of human multiple myeloma. Similar patterns emerged when the authors analyzed large patient datasets: individuals with DIS3 mutations were far more likely to carry IGH translocations and to show AID‑like mutation signatures in known myeloma driver genes, even though their overall mutation patterns across the genome looked similar to patients without DIS3 mutations.
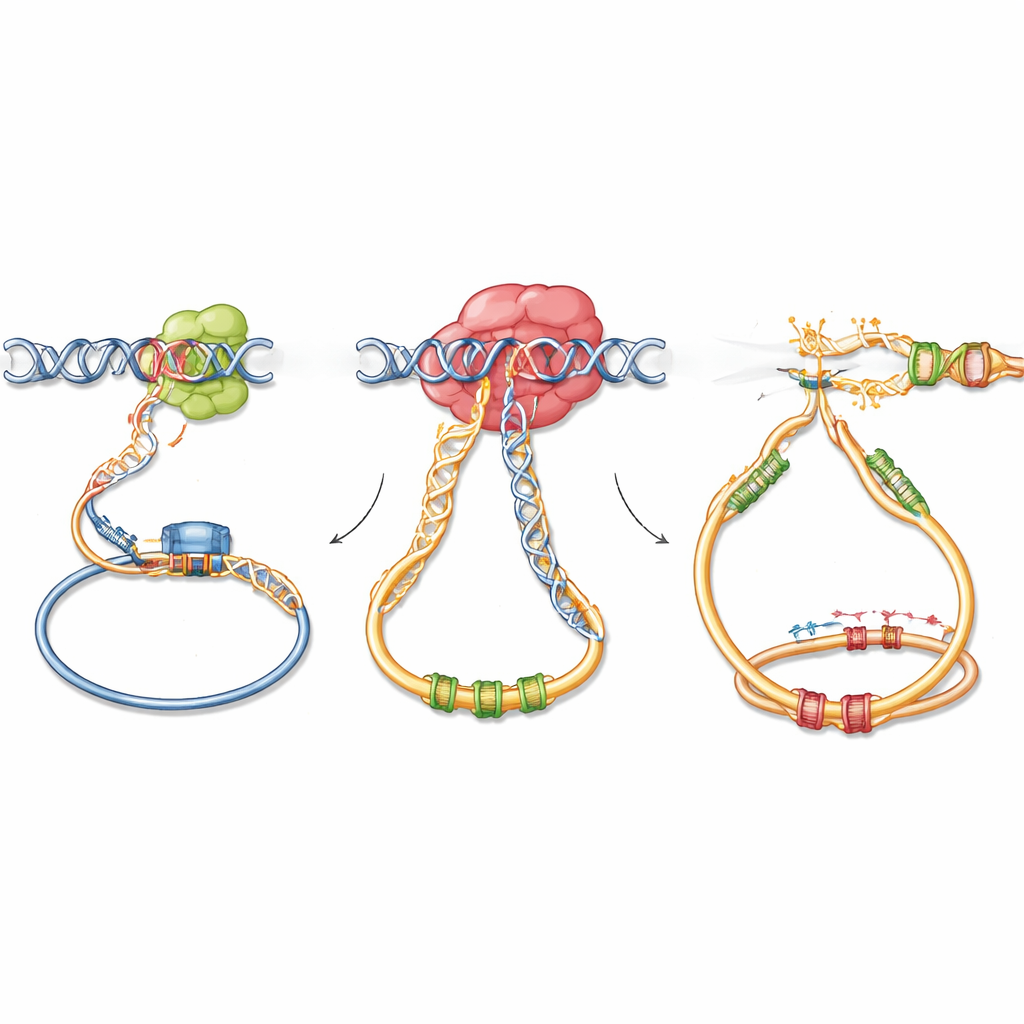
How Stalled RNA Cleanup Fuels DNA Damage
Mechanistic experiments revealed what goes wrong at the molecular level. The G766R version of DIS3 still works on simple RNAs but tends to stall on structured RNA and RNA–DNA hybrids close to active genes and regulatory regions. Using a sensitive mapping technique, the authors found that mutant DIS3 accumulates more strongly on chromatin at sites where AID is also recruited, especially around antibody switch regions, enhancer elements, and known AID off‑target genes. At these sites, RNA–DNA hybrids and unusual DNA structures build up, exposing stretches of single‑stranded DNA on both strands. In activated B cells and model cell lines, this led to more DNA damage foci, but only when AID was present. Importantly, the usual bias of AID toward one DNA strand was lost, producing more symmetrical damage on both strands, which favors staggered double‑strand breaks and error‑prone repair that stitches together mismatched DNA ends.
Hijacking Normal Genome Architecture
The study also asked whether the DIS3 mutation reshapes the 3D folding of DNA in the nucleus, which is crucial for bringing antibody genes and their regulatory elements together. High‑resolution chromosome conformation measurements showed that, unlike a full loss of DIS3, the G766R variant leaves the overall architecture of the antibody locus intact. Instead of rewiring the genome, the mutant DIS3 appears to exploit existing physical contacts: translocation breakpoints in mouse tumors cluster near active promoters, enhancers, and binding sites for architectural proteins that define chromatin loops. In effect, the mutation increases the chance that when AID‑induced breaks occur at these naturally neighboring sites, they will rejoin incorrectly, permanently linking strong antibody enhancers to nearby cancer‑promoting genes.
From Subtle Enzyme Change to Blood Cancer
Taken together, this work shows that multiple myeloma‑associated DIS3 mutations act not by broadly disrupting B‑cell biology, but by sharpening a very specific risk during short windows of B‑cell activation. The mutant enzyme lingers on structured RNAs at AID target sites, enhances AID’s access to both DNA strands, and raises the probability of misrepaired double‑strand breaks that fuse antibody regions to oncogenes. Because normal antibody switching and B‑cell responses remain largely intact, these “gain‑of‑function” DIS3 variants can persist long enough to seed the chromosomal rearrangements that drive multiple myeloma.
Citation: Kuliński, T.M., Gewartowska, O., Mahé, M. et al. DIS3 mutations enhance AID-driven translocations during B-cell activation, promoting transformation to multiple myeloma. Nat Commun 17, 3976 (2026). https://doi.org/10.1038/s41467-026-70386-3
Keywords: multiple myeloma, B cells, chromosomal translocations, RNA exosome, activation-induced cytidine deaminase