Clear Sky Science · sv
Järn‑boran‑katalyserad karbonylhydroborering och isolering av en järn(I)-ketylradikal
Att omvandla vanliga kemikalier till användbara alkoholer
Alkoholer finns överallt omkring oss, från läkemedel och parfymer till växtskyddsmedel. Många av dessa användbara molekyler framställs genom att omvandla "karbonyl"‑grupper — enkla kemiska enheter som förekommer i industriella råvaror och till och med i koldioxid — till alkoholer. Den här artikeln utforskar ett nytt sätt att göra det på med järn, en riklig och billig metall, tillsammans med en bor‑baserad partner, för att utföra dessa omvandlingar under relativt milda förhållanden samtidigt som en ovanlig, tidigare osedd reaktionsväg blottläggs.
Varför en ny väg spelar roll
Traditionella industriella metoder för att omvandla karbonylföreningar och koldioxid till alkoholer förlitar sig på vätgas under högt tryck eller hårda reaktionsförhållanden. Kemister söker efter mildare, mer selektiva vägar som ger mindre spill och som kan använda jordens rikliga metaller. Ett lovande angreppssätt är "hydroborering", där en bor‑väteenhet adderas över en kol‑syra dubbelbindning för att ge ett mellanled som kan omvandlas till en alkohol. Många katalysatorer baserade på sällsynta metaller är kända, men robusta järnbaserade system — och en klar bild av hur de fungerar — har saknats.
En järnpartner som gör det tunga arbetet
Författarna fokuserar på en tidigare utvecklad järnkomplex, kallad komplex A, som bär både fosfor och boratomer runt järncentret. De visar att detta komplex är en högst effektiv startkatalysator för hydroborering av ett brett spektrum ketoner (en huvudklass av karbonylföreningar), liksom ringslutna estrar kända som laktoner och till och med koldioxid. Med endast mycket små mängder av järnkomplexet och en vanlig borenhet (HBpin) omvandlas många ketoner vid rumstemperatur inom minuter till önskade borinnehållande mellanprodukter i höga utbyten. Systemet kan också utföra en sällsynt "dubbel hydroborering" på cykliska estrar, som effektivt öppnar ringen för att ge diol‑prekurser. För koldioxid möjliggör byte till en mer skrymmande borenhet, (9‑BBN)2, selektiv bildning av en metanol‑liknande produkt under milda förhållanden. 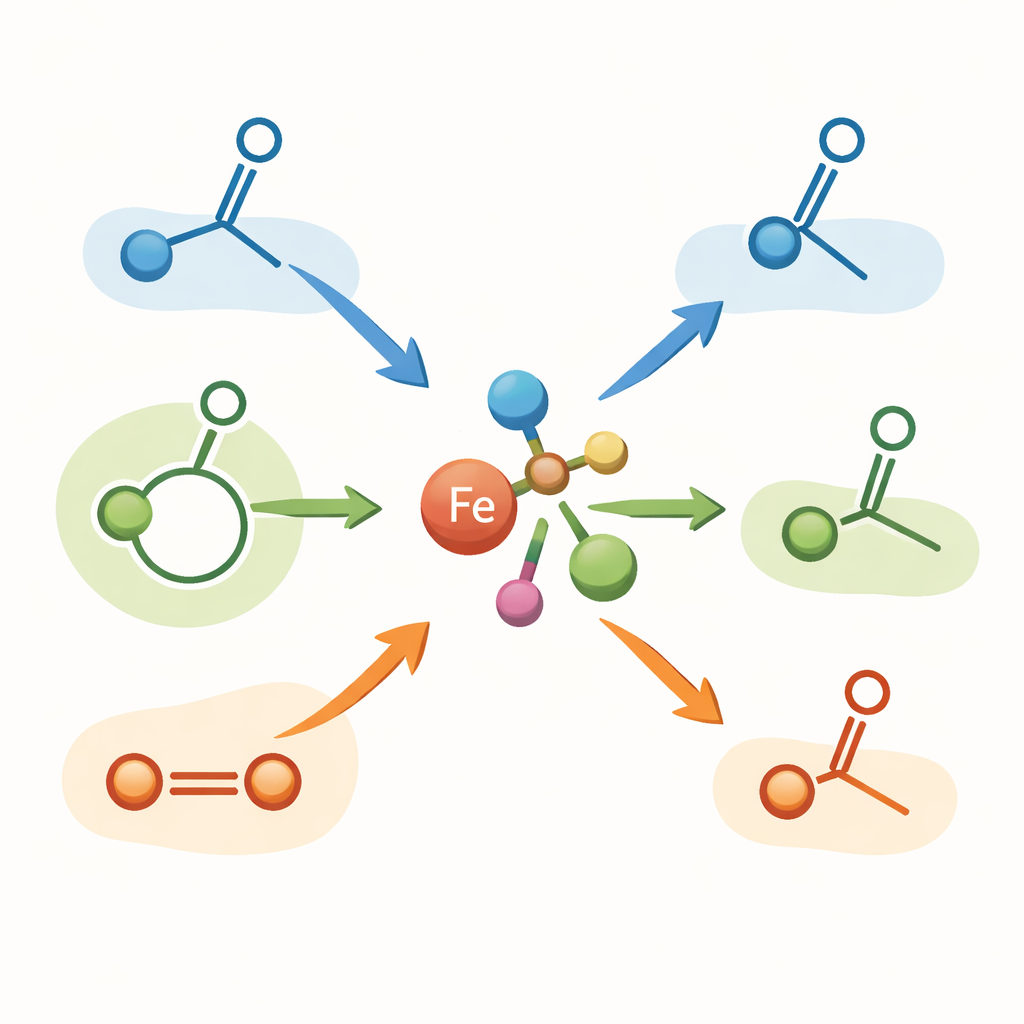
Att följa reaktionen i realtid
Utöver att demonstrera bred reaktivitet utreder forskarna hur järnkomplexet faktiskt driver reaktionen. Genom noggrann övervakning av reaktionshastigheter med hjälp av kärnmagnetisk resonansspektroskopi och genom att variera mängderna katalysator, keton och borenhet sluter de sig till att den aktiva järnarten beter sig som om endast en del av det dimeriska komplexet utför det katalytiska arbetet vid varje givet ögonblick. Hastigheten ökar med mer keton men planar så småningom ut, vilket tyder på att ett överskott av substrat kan binda upp järnet i inaktiva former. Förvånande nog saktar tillförsel av mer borenhet faktiskt ner reaktionen, vilket antyder att den kan konkurrera med ketonen om tillgång till katalysatorn. Isotopexperiment, där väte bundet till bor bytts mot dess tyngre kusin deuterium, visar att brytning av bor‑vätebindningen är ett viktigt, hastighetsbestämmande steg.
Att fånga en sällsynt radikal in action
För att se hur järnkomplexet ser ut när det möter en karbonylgrupp utförde teamet stökiometriska modellreaktioner. När komplex A möter bensofenon bildas en slående lila förening där ketonen har omvandlats till en "ketyl"‑radikal — i praktiken en karbonyl som plockat upp en extra elektron — och nu binder till järnet via sin syreatom. Detaljerade strukturella studier med röntgenkristallografi, magnetiska mätningar och beräkningar visar att denna art är ovanlig: den kombinerar ett järncenter med tre oparade elektroner och en ketylradikal som är magnetiskt motsatt dem, vilket ger ett totalt spinnläge på ett. Även om den är anmärkningsvärd visar ytterligare tester att denna radikalkomplex inte är huvudvägen för katalysen; istället sitter den sannolikt i jämvikt med mer konventionella två‑elektroniga mellanled. 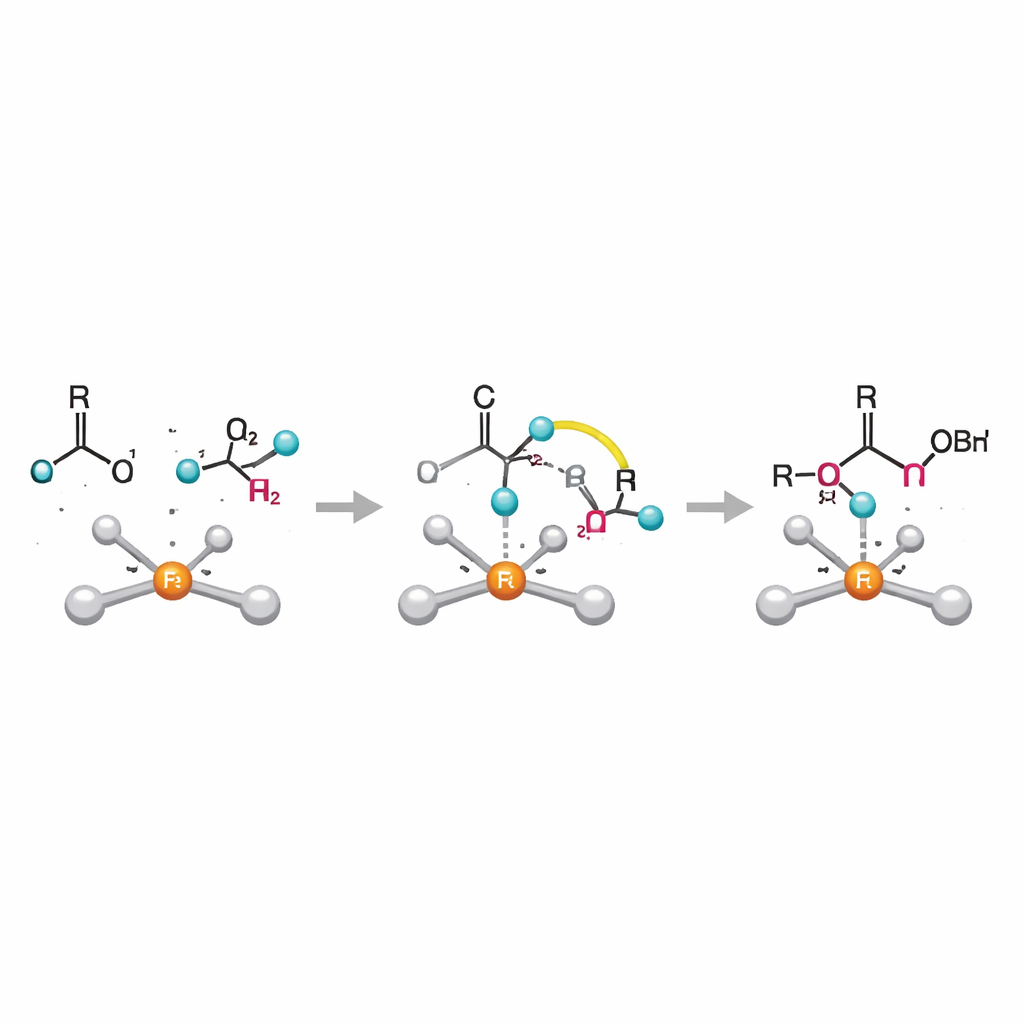
En ny sorts rörelse mellan partner
Med hjälp av kvantkemiska beräkningar kartlägger författarna en fullständig katalytisk cykel som stämmer med kinetiska och experimentella data. I deras bild binder borenheten först nära järncentret, och den inkommande ketonen kopplar sin syreatom till bor, vilket bildar en brygga mellan de två partnerna. Därefter kommer det avgörande steget: en direkt "ligand‑till‑ligand‑hydridöverföring", där väte som ursprungligen sitter på bor migrerar till kolatomen i ketonen medan hela händelsen dirigeras av järnet. Denna rörelse, snarare än ett traditionellt metallcentraliserat hydridskede, har inte tidigare rapporterats för denna typ av reaktion. När överföringen är slutförd lossnar det hydroborerade produkten och järnkomplexet reformeras, redo för en ny cykel.
Vad detta betyder framöver
I vardagliga termer visar detta arbete att ett smart utformat järnkomplex kan fungera som en dirigent, samordna atomrörelser mellan borenheter och karbonylgrupper för att göra alkoholprekursorer från vanliga kemikalier, inklusive koldioxid, under milda förhållanden. Samtidigt avslöjar det en ny sorts intern väte‑shuttling och en sällsynt järn‑ketylradikal, vilket fördjupar vår förståelse för hur sådana katalysatorer verkligen fungerar. Insikter som dessa kan vägleda designen av nästa generations lågkostnadssystem för att omvandla enkla, rikliga molekyler till värdefulla produkter mer effektivt och hållbart.
Citering: Grose, L.A., Schwamm, R.J., Brookfield, A. et al. Iron-borane catalyzed carbonyl hydroboration and isolation of an iron(I)-ketyl radical. Nat Commun 17, 2929 (2026). https://doi.org/10.1038/s41467-026-69500-2
Nyckelord: järnkatalys, hydroborering, karbonylreduktion, koldioxidkonvertering, ligand‑till‑ligand‑hydridöverföring