Clear Sky Science · sv
Hämning av PRMT5 försämrar Fanconi-anemi-vägens homologrekombination och ökar temozolomidets antitumöreffekt i glioblastom
Varför denna studie om hjärncancer är viktig
Glioblastom är en av de dödligaste hjärncancerformerna, och de flesta patienter får tillbaka sin tumör även efter operation, strålning och kemoterapi. Denna studie undersöker om en experimentell substans i kombination med standardkemoterapin temozolomid kan göra cancerceller mindre benägna att reparera sitt DNA, skjutsa dem mot självförstörelse och potentiellt ge patienterna längre överlevnadstid.
Hjärntumörer som reparerar sig själva alltför väl
Glioblastom uppträder inte som en enhetlig massa; det innehåller stamliknande celler som kan motstå behandling och starta ny tillväxt. Dessa celler är särskilt bra på att åtgärda DNA-skador, vilket dämpar effekten av strålning och läkemedel som temozolomid som verkar genom att skada DNA. Forskarna fokuserade på ett protein kallat PRMT5, som hjälper celler hantera DNA-reparation och finns i höga nivåer i glioblastom. Eftersom läkemedel som blockerar PRMT5 redan utvecklas, undrade teamet om att stänga av detta protein skulle göra tumörens stamliknande celler mindre kapabla att överleva temozolomidbehandling.
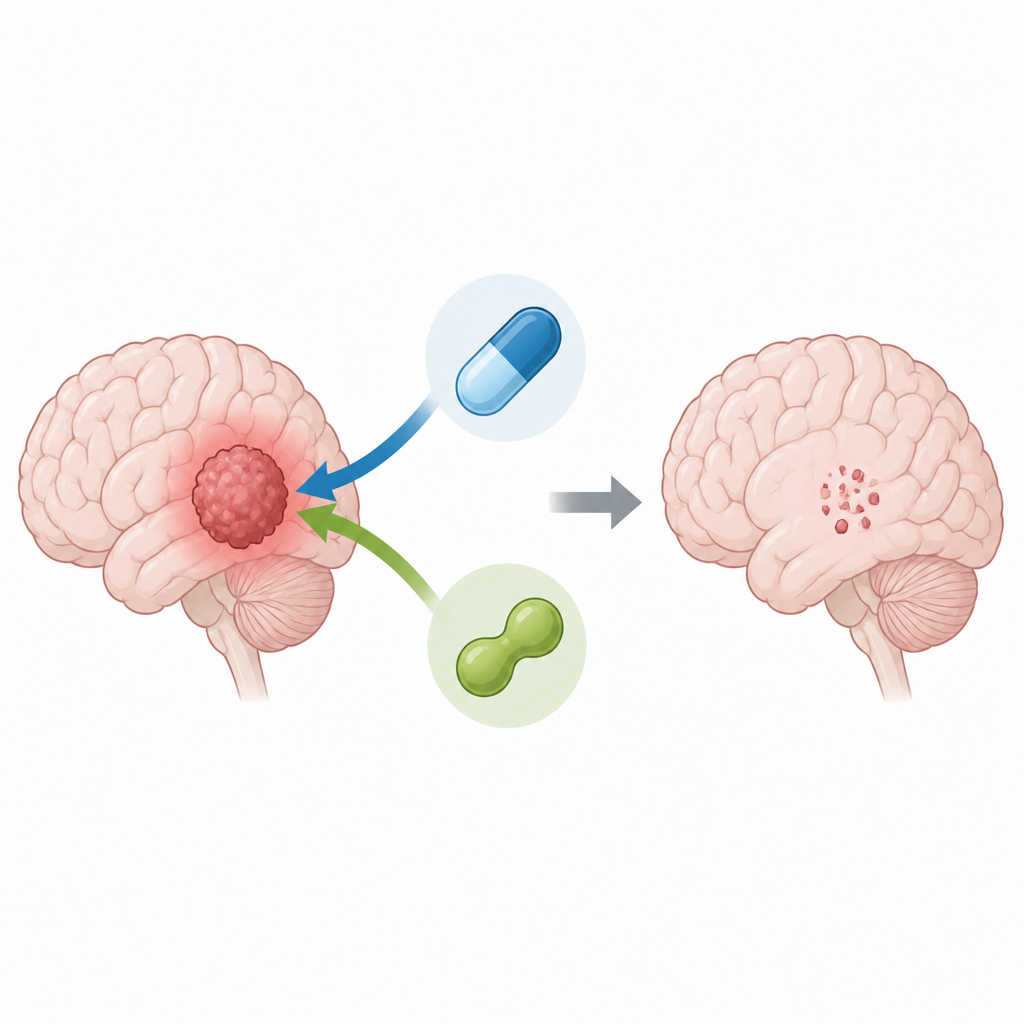
Kombinera två läkemedel för att slå hårdare mot tumörceller
Med patientavledda gliom-stamliknande celler odlad i labbet minskade forskarna antingen PRMT5-nivåerna med genetiska verktyg eller blockerade dess aktivitet med en förening kallad LLY-283. Därefter behandlade de cellerna med temozolomid. Celler med inaktiverat PRMT5 behövde mycket mindre temozolomid för att dödas, och datoranalys av läkemedelsresponser visade en tydlig synergistisk effekt, vilket betyder att de två behandlingarna tillsammans var kraftfullare än var och en för sig. Kombinationen minskade inte bara cellsurvival utan reducerade också dessa stamliknande cellers förmåga att bilda nya sfärer, en labbmetod för att mäta deras potential att återbilda tumörer.
Avväpna tumörens DNA-reparationsverktyg
För att förstå varför läkemedelsparet fungerade så väl undersökte teamet genaktivitet och viktiga reparationsproteiner. Blockering av PRMT5 sänkte aktiviteten hos många DNA-reparationsgener och försvagade särskilt en precis reparationsprocess kallad homolog rekombination. I kombination med temozolomid ledde PRMT5-hämning till fler DNA-brott i cancerceller, vilket syntes som ökade signaler för DNA-skada och längre ”komettailer” i ett standardtest där skadat DNA dras bort från cellkärnan. Utan tid att pausa i cellcykeln och laga skadorna ackumulerade tumörcellerna dödliga skador och drevs mot programmerad celldöd.
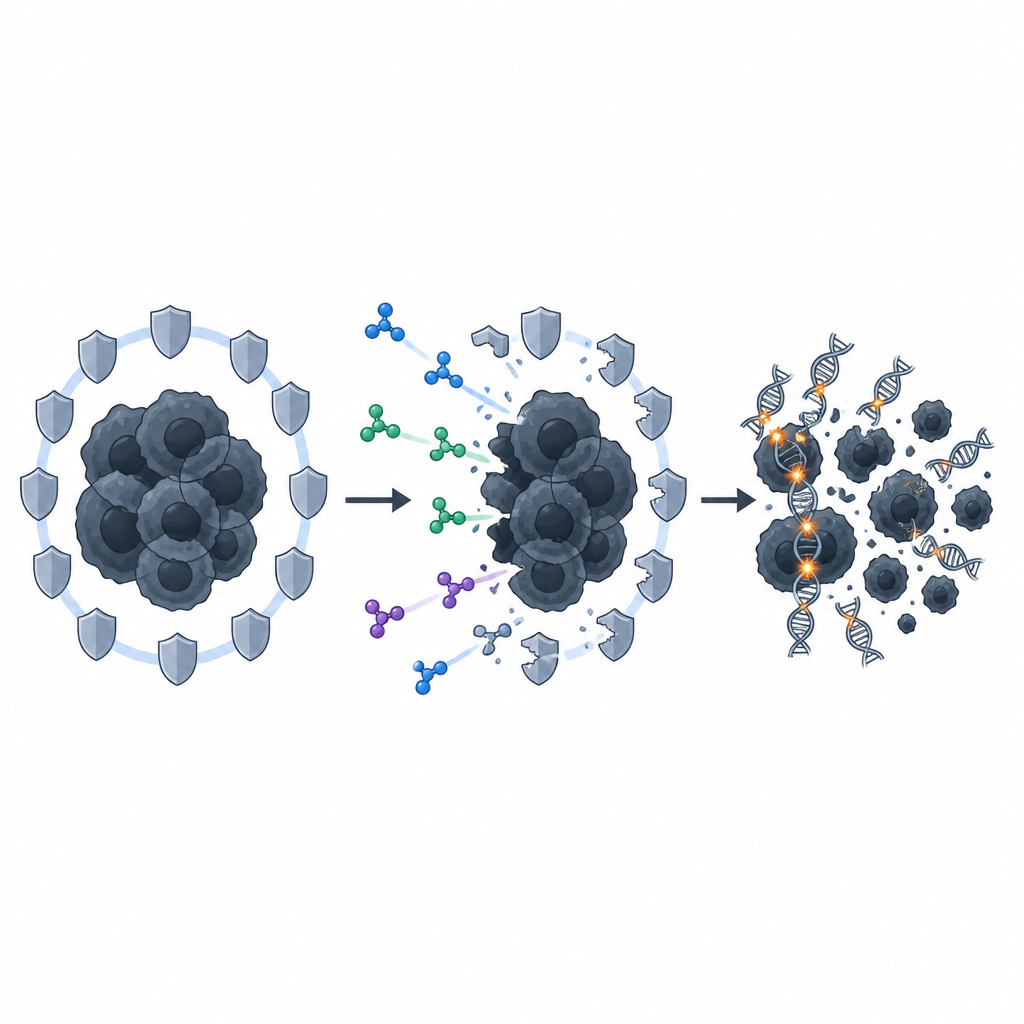
En tidigare dold hjälparväg träder fram
Vid närmare analys fann forskarna att en specifik reparationsväg kallad Fanconi-anemi-vägen starkt dämpades när PRMT5 blockeredes. Normalt aktiverar temozolomid gener i denna väg och ökar nivån av ett nyckelprotein, FANCD2, som i sin tur stödjer homolog rekombination via ett annat protein, RAD51. När PRMT5 hämnades klingade detta skyddande svar av: Fanconi-anemi-gener och FANCD2-nivåer sjönk, och RAD51-baserade reparationsstrukturer blev mindre frekventa. Att tysta FANCD2 direkt gav en liknande effekt och ökade känsligheten hos de stamliknande tumörcellerna för temozolomid genom att skära av deras förmåga att laga läkemedelsinducerade DNA-skador.
Testa strategin i djurmodeller
Teamet implantade sedan humana glioblastom-stamliknande celler i hjärnorna på möss för att efterlikna sjukdomen hos människor. Möss som behandlades med antingen temozolomid ensamt eller PRMT5-hämning ensamt överlevde något längre än obehandlade djur, men de som fick kombinationen levde märkbart längre och hade långsammare tumörtillväxt. Vävnadsanalyser från dessa möss visade färre delande celler och fler tecken på DNA-brott och celldöd i tumörer som exponerats för båda behandlingarna, i linje med idén att blockering av PRMT5 lämnar cancerceller försvarslösa mot temozolomidinducerad skada.
Vad detta kan betyda för framtida patienter
Enkelt uttryckt visar denna studie att glioblastomceller är beroende av PRMT5 och Fanconi-anemi-vägen för att reparera DNA-skador orsakade av temozolomid och undkomma destruktion. Genom att stänga av PRMT5 med ett läkemedel som LLY-283 kan forskare försvaga dessa reparationssystem, så att standardkemoterapin fungerar mer effektivt och möjligen i lägre doser. Även om arbetet fortfarande befinner sig i preklinisk fas beskriver det en tydlig strategi: gör envisa hjärntumörer mindre kapabla att laga sitt eget DNA, så har befintliga behandlingar större chans att hålla sjukdomen i schack.
Citering: Onishi, S., Jayamohan, S., Chowdhury, A. et al. PRMT5 inhibition impairs Fanconi Anemia pathway-mediated homologous recombination and enhances the antitumor efficacy of Temozolomide in glioblastoma. Cell Death Dis 17, 505 (2026). https://doi.org/10.1038/s41419-026-08739-5
Nyckelord: glioblastom, temozolomid, DNA-reparation, PRMT5-hämmare, Fanconi-anemi-vägen