Clear Sky Science · en
PRMT5 inhibition impairs Fanconi Anemia pathway-mediated homologous recombination and enhances the antitumor efficacy of Temozolomide in glioblastoma
Why this brain cancer study matters
Glioblastoma is one of the deadliest brain cancers, and most patients see their tumors return even after surgery, radiation, and chemotherapy. This study explores whether pairing an experimental drug with the standard chemotherapy temozolomide can make cancer cells less able to repair their DNA, pushing them toward self-destruction and potentially offering patients more time.
Brain tumors that repair themselves too well
Glioblastoma does not behave like a uniform mass; it contains stem-like cells that can resist treatment and seed new growth. These cells are especially good at fixing DNA damage, which blunts the effects of radiation and drugs like temozolomide that work by harming DNA. The researchers focused on a protein called PRMT5, which helps cells manage DNA repair and is found at high levels in glioblastoma. Because drugs that block PRMT5 are already in development, the team asked whether shutting down this protein would make tumor stem-like cells less able to survive temozolomide.
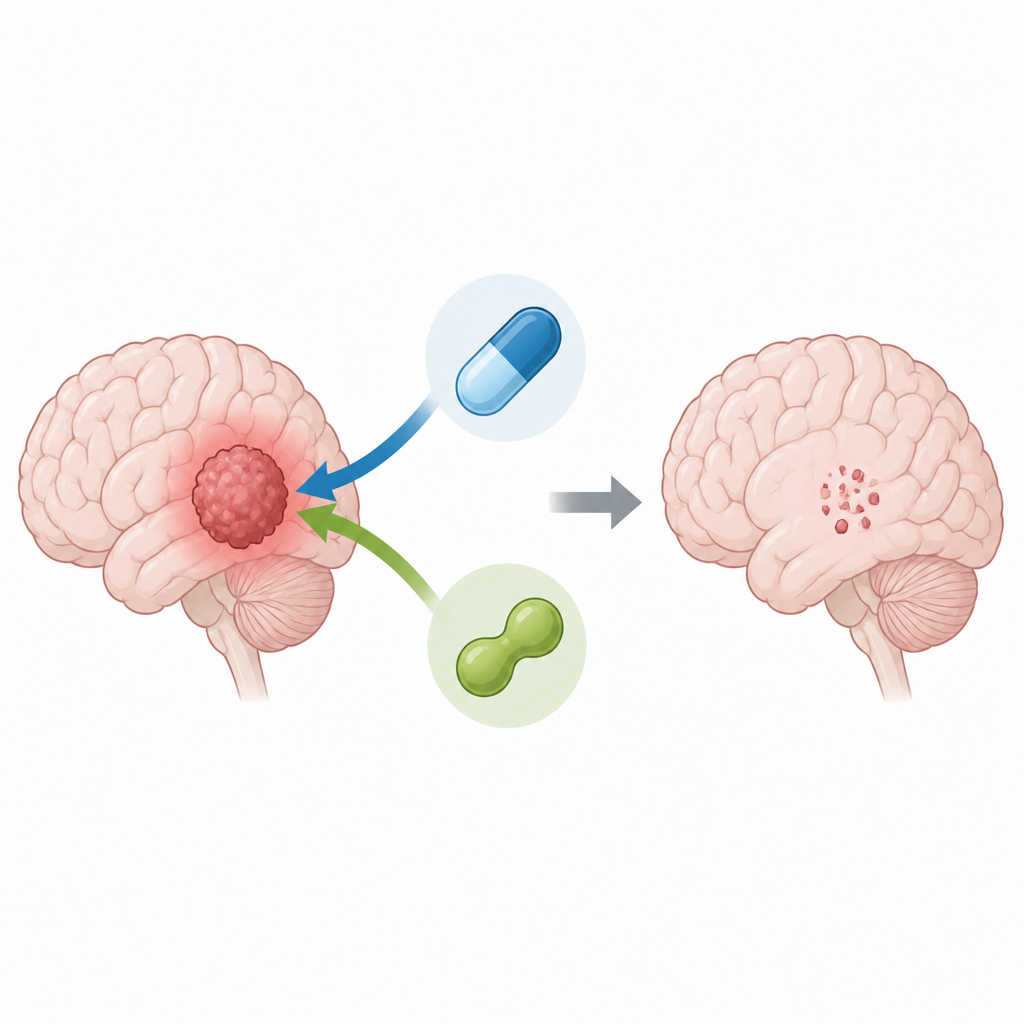
Pairing two drugs to hit tumor cells harder
Using patient-derived glioma stem-like cells grown in the lab, the scientists either reduced PRMT5 levels with genetic tools or blocked its activity with a compound called LLY-283. They then treated the cells with temozolomide. Cells with disabled PRMT5 needed far less temozolomide to be killed, and computer analysis of the drug responses showed a clear synergistic effect, meaning the two treatments together were more powerful than either alone. The combination not only reduced cell survival but also cut down the ability of these stem-like cells to form new spheres, a lab measure of their potential to regrow tumors.
Disarming the tumor’s DNA repair toolkit
To understand why the drug pair worked so well, the team examined patterns of gene activity and key repair proteins. Blocking PRMT5 reduced the activity of many DNA repair genes and, in particular, weakened a precise repair process called homologous recombination. When combined with temozolomide, PRMT5 inhibition led to more DNA breaks in cancer cells, seen as increased DNA damage signals and longer “comet tails” in a standard assay where damaged DNA trails away from the cell nucleus. Without time to pause in the cell cycle and fix this damage, the tumor cells accumulated lethal injuries and were driven toward programmed cell death.
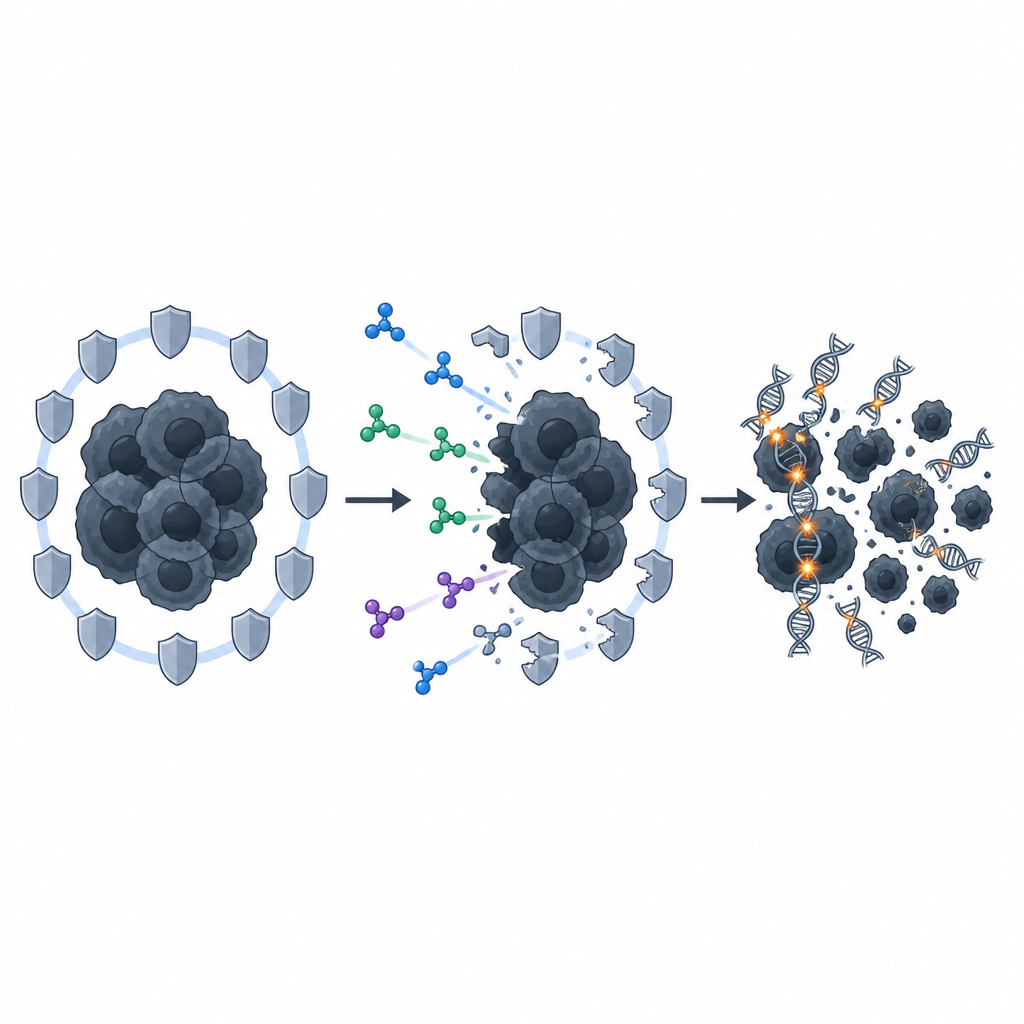
A hidden helper pathway comes into view
Digging deeper, the researchers found that a specific repair route called the Fanconi anemia pathway was strongly dampened when PRMT5 was blocked. Normally, temozolomide switched on genes in this pathway and boosted a key protein, FANCD2, which in turn supported homologous recombination through another protein, RAD51. When PRMT5 was inhibited, this protective response was blunted: Fanconi anemia genes and FANCD2 levels dropped, and RAD51 repair structures became less abundant. Silencing FANCD2 directly had a similar effect, further sensitizing the tumor stem-like cells to temozolomide by cutting off their ability to patch up drug-induced DNA damage.
Putting the strategy to the test in animals
The team then implanted human glioblastoma stem-like cells into the brains of mice to mimic the human disease. Mice treated with either temozolomide alone or PRMT5 inhibition alone survived slightly longer than untreated animals, but those that received the combination lived noticeably longer and had slower-growing tumors. Tissue analysis from these mice revealed fewer dividing cells and more signs of DNA breaks and cell death in tumors exposed to both treatments, consistent with the idea that blocking PRMT5 leaves cancer cells defenseless against temozolomide-induced damage.
What this could mean for future patients
In simple terms, this study shows that glioblastoma cells rely on PRMT5 and the Fanconi anemia pathway to repair the DNA damage caused by temozolomide and escape destruction. By turning off PRMT5 with a drug like LLY-283, researchers can weaken these repair systems, allowing standard chemotherapy to work more effectively and at potentially lower doses. While this work is still at the preclinical stage, it outlines a clear strategy: make stubborn brain tumors less capable of fixing their own DNA, so existing treatments have a better chance to keep the disease at bay.
Citation: Onishi, S., Jayamohan, S., Chowdhury, A. et al. PRMT5 inhibition impairs Fanconi Anemia pathway-mediated homologous recombination and enhances the antitumor efficacy of Temozolomide in glioblastoma. Cell Death Dis 17, 505 (2026). https://doi.org/10.1038/s41419-026-08739-5
Keywords: glioblastoma, temozolomide, DNA repair, PRMT5 inhibitor, Fanconi anemia pathway