Clear Sky Science · pt
Analisando o transporte de calor em polímeros cristalinos no espaço real e no espaço recíproco
Por que o calor em plásticos pode viajar como em metais
A maioria de nós pensa em plásticos como bons isolantes térmicos, mas quando suas cadeias se alinham perfeitamente, alguns plásticos podem conduzir calor quase tão bem quanto metais. Este artigo explora como o calor se move através de formas altamente ordenadas de dois polímeros comuns—polietileno (usado em plásticos do dia a dia) e politiofeno (um polímero semicondutor modelo)—e faz uma pergunta aparentemente simples: métodos de simulação muito diferentes podem concordar sobre quão bem esses materiais conduzem calor?
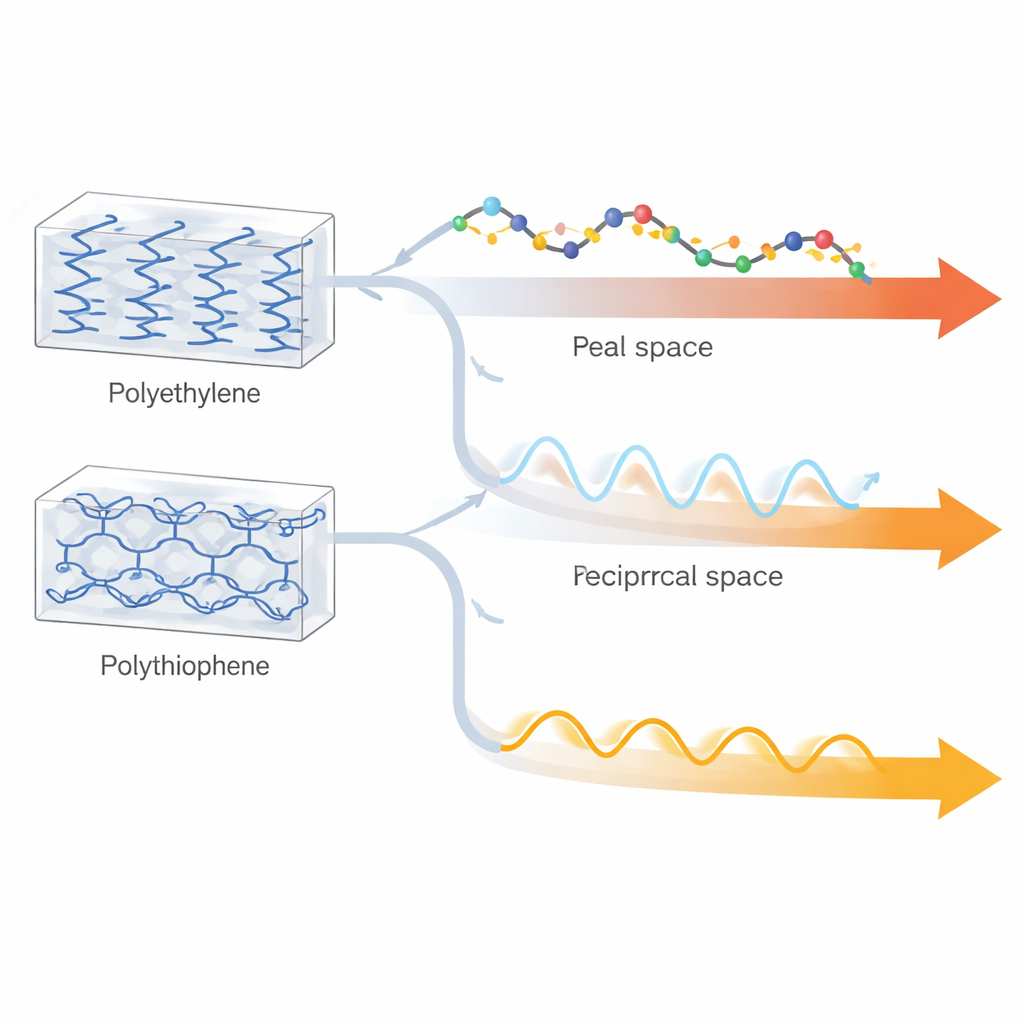
Dois plásticos organizados com personalidades muito diferentes
O estudo foca no polietileno e no politiofeno cristalinos, onde longas cadeias moleculares são empacotadas em arranjos repetitivos e bem ordenados. Em seu estado habitual, emaranhado e amorfo, esses polímeros mal conduzem calor, mas quando as cadeias são esticadas e alinhadas, medições em fibras e filmes de polietileno mostram condutividades térmicas que rivalizam com alguns metais na direção da cadeia. Para o politiofeno, existiam apenas dados teóricos. Conhecer o limite superior real do fluxo de calor em um cristal perfeitamente ordenado é crucial para projetar dissipadores de calor leves e eletrônicos flexíveis avançados, ainda que cálculos anteriores para o polietileno tenham discordado por fatores de vários, dependendo do método e dos modelos de interação usados.
Duplas maneiras de observar o movimento do calor
Os autores comparam duas famílias amplas de abordagens. Em simulações de “espaço real”, a dinâmica molecular acompanha o movimento de átomos individuais ao longo do tempo: aplica-se uma diferença de temperatura, observa-se o fluxo de energia e extrai-se a condutividade térmica. Em abordagens do “espaço recíproco”, o mesmo processo é descrito em termos de fônons—ondas vibracionais quantizadas—cuja velocidade, vida útil e ocupação determinam o transporte de calor via equação de transporte de Boltzmann. Cada abordagem tem compromissos embutidos: cálculos baseados em fônons geralmente incluem apenas os eventos de espalhamento mais simples entre três fônons, mas tratam corretamente a estatística quântica; a dinâmica molecular inclui naturalmente todos os níveis de anarmonicidade (espalhamentos complexos), porém se apoia em estatística clássica, que se torna questionável para vibrações de alta frequência à temperatura ambiente.
Aprendizado de máquina como língua comum
Um passo central para tornar esses métodos comparáveis é como as forças atômicas são calculadas. Em vez de confiar em campos de força tradicionais, muitas vezes imprecisos, ou em cálculos quânticos proibitivamente caros a cada passo, os pesquisadores usam potenciais “tensor de momento” aprendidos por máquina. Estes são treinados em um conjunto limitado de dados quântico-mecânicos de alta precisão e então usados para executar simulações muito longas e muito grandes com fidelidade próxima à de primeiro princípio. A equipe constrói deliberadamente versões ligeiramente diferentes desses potenciais, otimizadas quer para propriedades vibracionais precisas quer para dinâmica molecular estável a longo prazo, e verifica cruzadamente que a variação nos resultados permanece pequena em comparação com as tendências físicas que desejam resolver.
Quando tudo funciona bem: o caso do politiofeno
Para o politiofeno cristalino, todas as abordagens convergem para quase a mesma resposta. Cálculos baseados em fônons que incluem apenas espalhamento de três fônons preveem condutividades térmicas na direção da cadeia de aproximadamente 80–100 W m−1 K−1, dependendo de se é usada uma simplificação padrão ou se é resolvida uma equação mais completa. Rotas baseadas em dinâmica molecular—tanto as que extraem tempos de vida de fônons a partir de trajetórias quanto os métodos totalmente em espaço real que impõem ou relaxam gradientes de temperatura—caem essencialmente na mesma faixa uma vez aplicadas correções pequenas e bem compreendidas. Um exame mais atento revela por quê: os principais portadores de calor são vibrações de frequência relativamente baixa, para as quais as estatísticas clássicas e quânticas são bastante similares à temperatura ambiente, e processos de três fônons já oferecem meios suficientes para o espalhamento de energia. Neste polímero, portanto, os diferentes métodos são consistentes e as aproximações de cada lado causam pouco dano.
Quando a simplicidade complica: o caso do polietileno
O polietileno comporta-se de forma muito diferente. Sua espinha simples e repetitiva deixa menos ramos vibracionais, e as regras de conservação de energia e momento suprimem muitos canais de espalhamento de três fônons em uma banda de modos de frequência mais alta entre cerca de 11 e 16 terahertz. Em cálculos padrão de fônons que incluem apenas processos de três fônons, esses modos adquirem tempos de vida extraordinariamente longos e dominam o transporte de calor, produzindo condutividades previstas impressionantemente altas acima de 300 W m−1 K−1. Quando os autores, em vez disso, inferem tempos de vida de fônons a partir de dinâmica molecular—onde todo espalhamento de ordem superior está presente—esses mesmos modos ainda importam, mas seus tempos de vida encolhem dramaticamente, reduzindo a condutividade em mais de um fator de dois. Como esses modos importantes estão em altas frequências, a estatística clássica também começa a falhar, e usar uma descrição clássica versus quântica de suas populações muda a resposta em quase 50 por cento.
O que isso significa para projetar plásticos que conduzem calor
Ao combinar forças precisas aprendidas por máquina com um conjunto de métodos complementares, o estudo mostra que descrições consistentes do transporte de calor em polímeros cristalinos são alcançáveis—mas somente se a física sutil de cada material for respeitada. Para o politiofeno, o espalhamento de três fônons mais qualquer uma das estratégias de simulação comuns já fornece um quadro confiável. Para o polietileno, entretanto, os mesmos atalhos superestimam seriamente quão bem um cristal perfeito pode conduzir calor, porque falham em captar ou tratam mal tanto o espalhamento de ordem superior quanto a natureza quântica dos modos vibracionais de alta frequência. Os autores concluem que tentativas futuras de projetar fibras e filmes poliméricos de condutividade ultraelevada devem levar em conta esses efeitos se quiserem metas realistas, e que verificar cruzadamente as abordagens em espaço real e recíproco é uma forma eficaz de expor suposições ocultas em modelos de transporte térmico.
Citação: Reicht, L., Legenstein, L., Wieser, S. et al. Analysing heat transport in crystalline polymers in real and reciprocal space. npj Comput Mater 12, 129 (2026). https://doi.org/10.1038/s41524-026-01988-0
Palavras-chave: polímeros cristalinos, condutividade térmica, fônons, dinâmica molecular, potenciais de aprendizado de máquina