Clear Sky Science · pt
Mudanças no sistema inibitório GABAérgico cortical em um modelo murino de Atrofia Muscular Espinhal
Por que o equilíbrio cerebral importa em uma doença muscular
A Atrofia Muscular Espinhal (AME) é mais conhecida como uma doença pediátrica devastadora que enfraquece os músculos e pode ser fatal. Durante anos, os cientistas se concentraram principalmente nos neurônios motores da medula espinhal que controlam diretamente o movimento. Este estudo faz uma pergunta diferente: e se parte do problema também estiver mais acima, no próprio centro de movimento do cérebro? Ao investigar como as células “freio” no córtex motor funcionam mal em um modelo murino severo de AME, os pesquisadores revelam uma camada oculta da doença que pode ajudar a explicar sintomas e apontar novas estratégias terapêuticas.
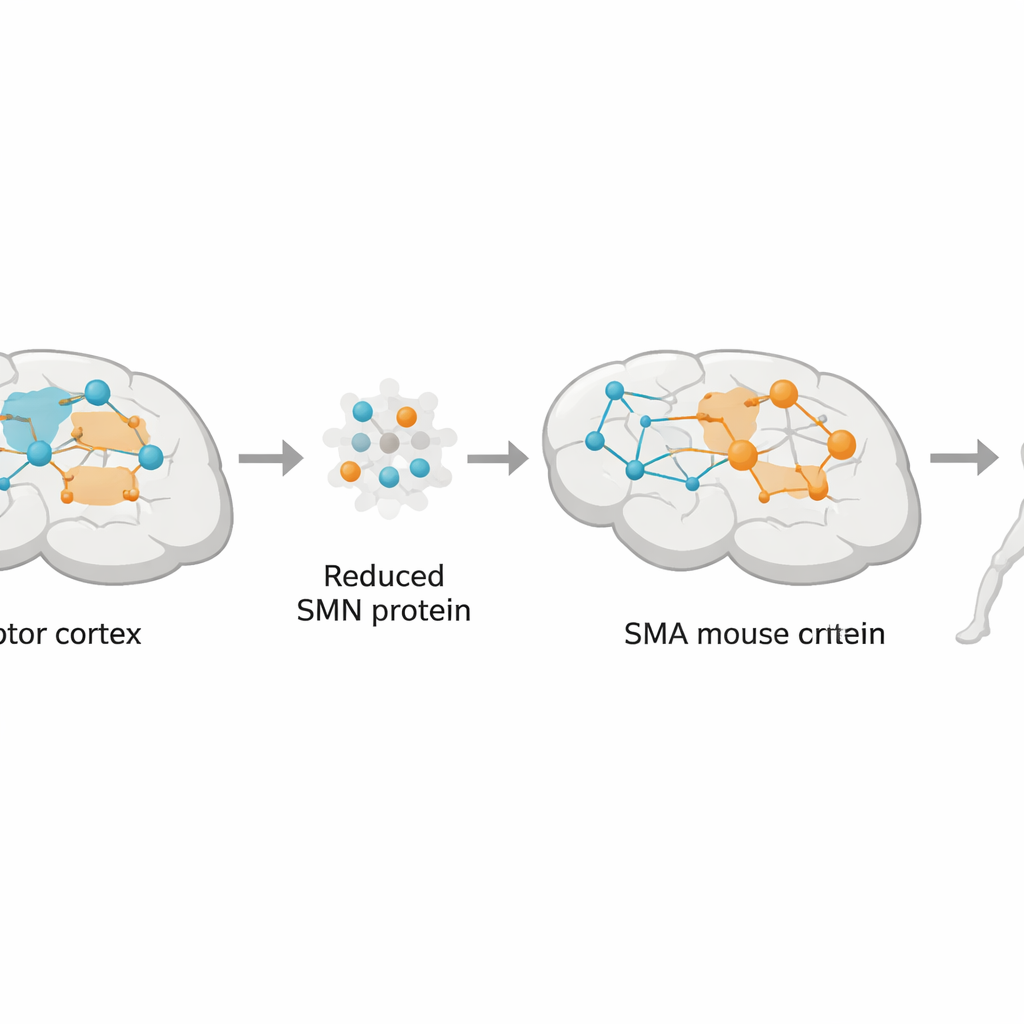
Uma trégua frágil entre sinais cerebrais
O movimento normal depende de uma trégua entre dois tipos de atividade celular no córtex: sinais excitatórios que levam os neurônios a disparar e sinais inibitórios que os contêm. Muitas doenças cerebrais, da epilepsia ao Parkinson, são hoje entendidas como perturbações desse equilíbrio excitatório–inibitório. Na AME, pacientes e modelos animais mostram alterações estruturais e funcionais no córtex motor, sugerindo que o “sistema de freio” também pode estar alterado ali. Ainda assim, a maior parte da pesquisa concentrou-se na medula espinhal. Os autores propuseram testar se a sinalização inibitória no córtex sensório-motor está perturbada na AME e se essa perturbação está ligada à perda de uma proteína chamada SMN, cuja deficiência causa a doença.
As células de freio do cérebro sob estresse
Usando uma combinação de imagens cerebrais, ensaios moleculares e registros elétricos, a equipe examinou o córtex sensório-motor de camundongos com AME em diferentes estágios da doença. Eles focalizaram no GABA, o principal mensageiro inibitório do cérebro, e em uma classe chave de células produtoras de GABA conhecidas como interneurônios parvalbumina, que atuam como freios rápidos e precisos na saída motora. Em camundongos com AME em estágio avançado, a densidade de neurônios positivos para GABA e a intensidade do sinal de GABA foram reduzidas, especialmente na camada profunda 5 do córtex, onde residem neurônios de saída que comandam a medula espinhal. As enzimas que sintetizam GABA (GAD65 e GAD67) também diminuíram, e os interneurônios parvalbumina exibiram menos ramificações e corpos celulares menores, sugerindo perda de força inibitória exatamente onde isso é mais importante para controlar o movimento.
Sinapses mais fracas e química desajustada
Para ver como essas alterações estruturais afetam a função, os pesquisadores mediram correntes elétricas inibitórias recebidas por neurônios piramidais da camada 5. Nos camundongos com AME, essas células experimentaram menos eventos inibitórios desencadeados por potenciais de ação, embora o tamanho de cada evento fosse maior — um padrão consistente com um sistema de freio em falha, mas parcialmente compensatório. A análise microscópica confirmou menos contatos sinápticos inibitórios nessas células, tanto em tecido cerebral quanto em culturas celulares simplificadas. Ao mesmo tempo, o perfil químico do córtex revelou um acúmulo de glutamina em estágio avançado, um precursor usado pelos neurônios para produzir tanto glutamato quanto GABA. Em vez de uma simples escassez de GABA no tecido como um todo, esses achados apontam para um redirecionamento inadequado do ciclo glutamina–glutamato–GABA dentro de circuitos locais.
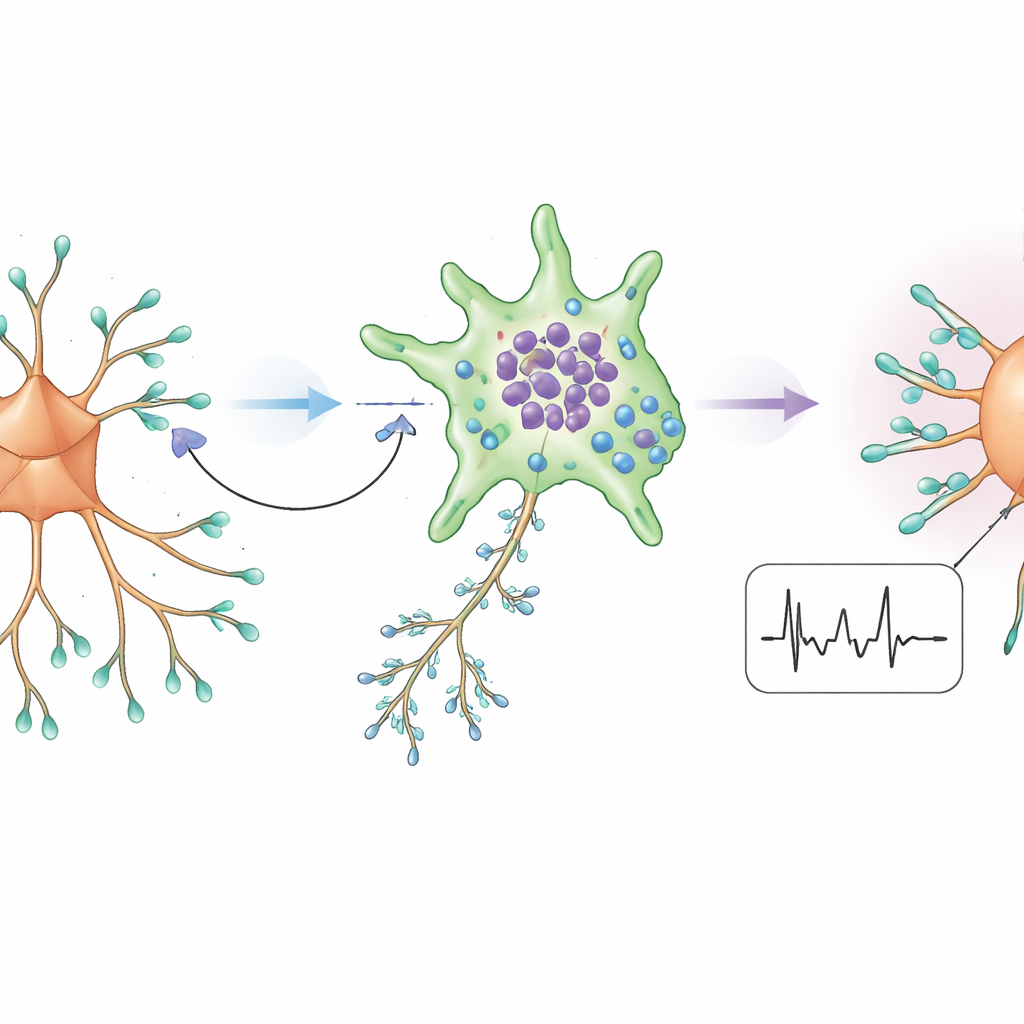
Astrócitos, transportadores e a conexão com SMN
Como a proteína SMN ajuda a regular o processamento de RNA e, assim, a produção de proteínas, a equipe investigou como sua perda poderia distorcer esse ciclo químico. Eles descobriram que um transportador de glutamina predominantemente produzido por astrócitos — um tipo de célula de suporte que fornece combustível aos neurônios — estava reduzido no córtex de AME. Outro transportador que reabsorve GABA para dentro dos astrócitos estava aumentado, e astrócitos em culturas de AME acumularam mais sinal semelhante ao GABA do que neurônios vizinhos. Quando os níveis de SMN foram experimentalmente reduzidos em neurônios que, de outra forma, eram normais, seu sinal de GABA diminuiu; quando o SMN foi aumentado com o fármaco para AME Nusinersen em cultura, o transporte de glutamina e os níveis de GABA melhoraram. Juntos, esses resultados sugerem que a deficiência de SMN interrompe a cooperação neurônio–astrócito, privando neurônios inibitórios das matérias-primas de que precisam enquanto encoraja astrócitos a sequestrar mais do GABA disponível.
O que isso significa para pessoas com AME
Para um leitor leigo, a mensagem é que a AME não é apenas uma doença da medula espinhal, mas também de circuitos cerebrais que moldam o movimento. O estudo mostra que, em um modelo murino severo de AME, o córtex motor gradualmente perde parte de seu sistema de freio: células inibitórias especializadas encolhem, produzem menos GABA, formam menos sinapses e são prejudicadas por um suporte defeituoso de astrócitos próximos. Isso deixa os neurônios de saída mais vulneráveis e pode agravar os sintomas motores mesmo quando as terapias atuais que aumentam SMN prolongam a sobrevida. O trabalho ajuda a explicar por que drogas que reforçam a sinalização inibitória às vezes mostraram benefícios na AME e sugere que tratamentos futuros podem precisar combinar a restauração de SMN com estratégias que estabilizem diretamente a inibição cortical e o metabolismo neurônio–astrócito.
Citação: Menduti, G., Ferrini, F., Caretto, A. et al. Changes in the cortical GABAergic inhibitory system in a Spinal Muscular Atrophy mouse model. Cell Death Dis 17, 285 (2026). https://doi.org/10.1038/s41419-026-08520-8
Palavras-chave: atrofia muscular espinhal, córtex motor, inibição GABA, interneurônios, metabolismo neurônio–astrócito