Clear Sky Science · pl
HarveST wykorzystuje heterogeniczne uczenie grafowe do ujawniania wzorców w przestrzennej transkryptomice
Widzieć, gdzie geny działają w tkankach
Nasze ciała zbudowane są z sąsiedztw komórek, z których każde pełni różne funkcje — od myślenia w mózgu po zwalczanie guzów w nowotworach. Rosnąca technologia zwana przestrzenną transkryptomiką pozwala naukowcom odwzorować, które geny są aktywne w różnych miejscach przekroju tkanki. Przekształcenie tych surowych map w czytelne, znaczące regiony i listy istotnych genów wciąż jednak bywa trudne. W tym badaniu przedstawiono nową metodę komputerową, HarveST, która pomaga badaczom wyraźniej zobaczyć strukturę tkanki i jej kluczowe geny, nawet w złożonych organach i guzach.
Dlaczego mapowanie sąsiedztw komórkowych jest trudne
W tkankach pobliskie komórki często działają wspólnie jako funkcjonalne wspólnoty, takie jak warstwy w mózgu czy strefy wokół guza. Przestrzenna transkryptomika mierzy aktywność genów w tysiącach drobnych punktów na przekroju tkanki, wraz z ich dokładnymi położeniami. Istniejące narzędzia komputerowe próbują grupować te punkty w domeny przestrzenne — regiony o podobnej aktywności genów i bliskim sąsiedztwie. Wiele metod albo ignoruje położenie przestrzenne, albo zakłada, że sąsiednie punkty muszą należeć do tego samego regionu. Może to rozmazać ważne granice, przeoczyć rzadkie skupiska komórek i nie powiązać każdego regionu z genami, które go rzeczywiście definiują.
Nowy sposób łączenia przestrzeni i genów
HarveST rozwiązuje te problemy, przekształcając każdy zbiór danych w bogatą sieć, która jednocześnie łączy miejsca tkankowe i geny 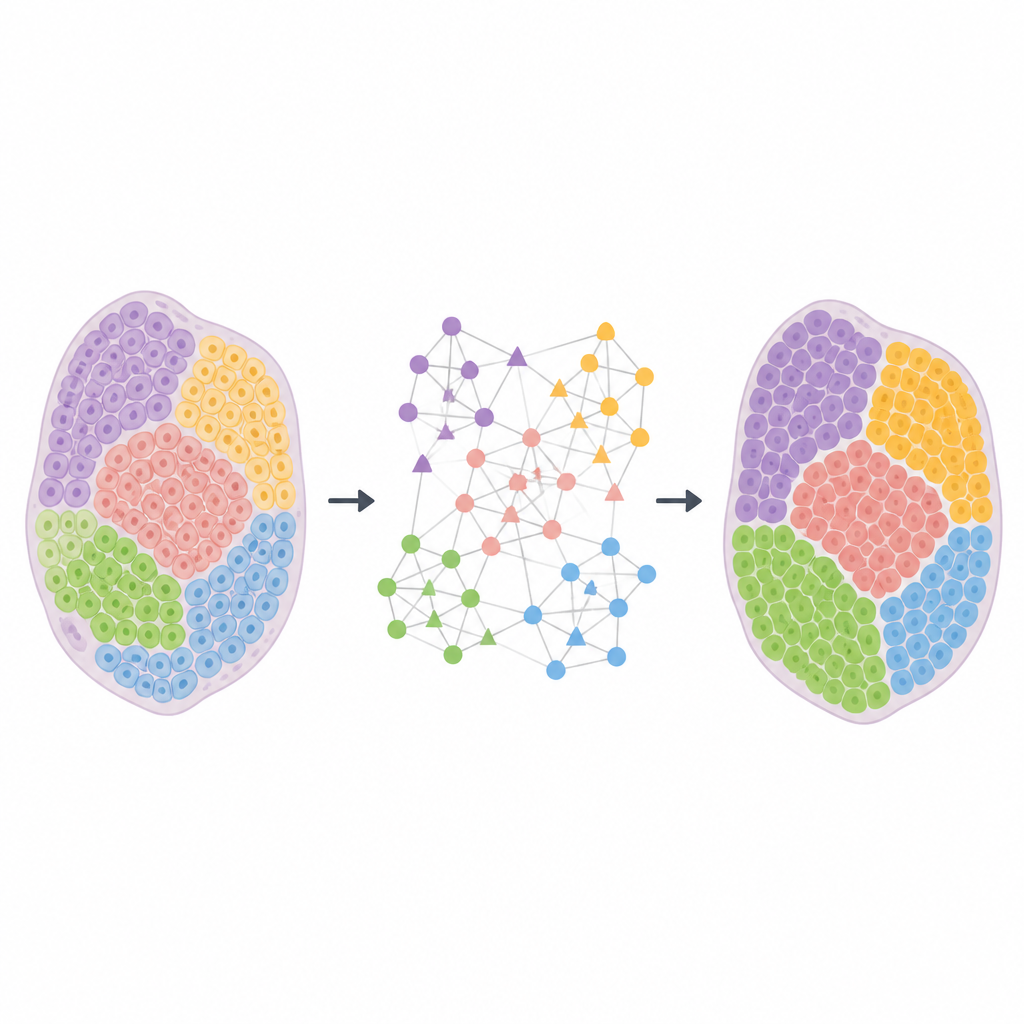
Znajdowanie właściwych genów dla każdego regionu
Ponad samo rysowanie map regionów tkankowych, HarveST ma na celu znalezienie genów markerowych, które czynią każdy region wyjątkowym. Zamiast testować każdy gen osobno, metoda uruchamia „losowy spacer” z miejsc wewnątrz wybranego regionu i pozwala temu sygnałowi rozprzestrzeniać się przez sieć 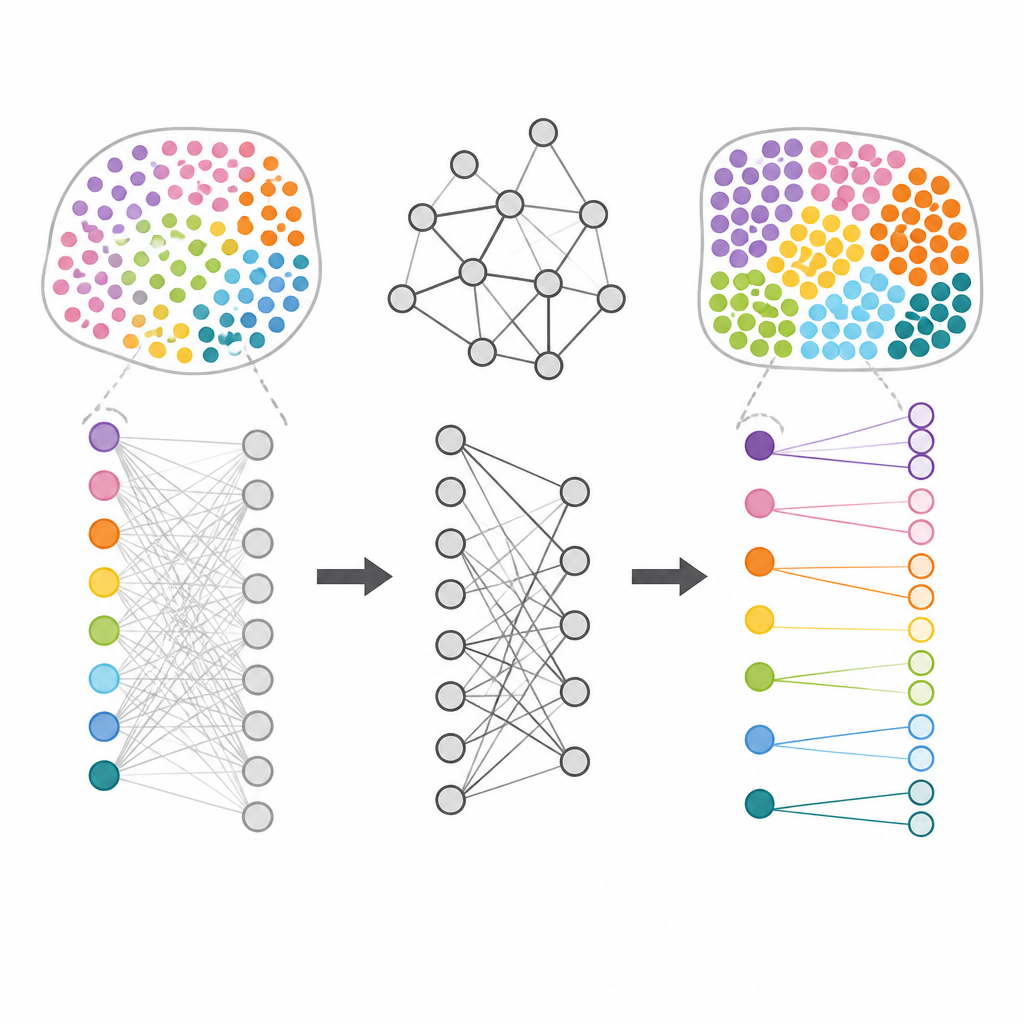
Co HarveST ujawnia w mózgach i guzach
Aby przetestować HarveST, zespół zastosował go do próbek ludzkiego mózgu, raka piersi, raka trzustki oraz mysiego opuszki węchowej mierzonych na kilku platformach przestrzennej transkryptomiki. W ludzkiej korze przedczołowej HarveST dokładniej odtworzył klasyczne sześć warstw kory i istotę białą niż czternaście konkurencyjnych metod, osiągając wyższe wyniki na szerokim zestawie metryk klastrowania i granic. W danych o wysokiej rozdzielczości z mysiej opuszki węchowej wyróżnił się oddzielając cienkie, gęsto upakowane warstwy, które inne narzędzia łączyły ze sobą. W rakach piersi i trzustki HarveST lepiej rozróżniał krawędzie guza, regiony normalne i strefy inwazyjnego nowotworu, łącząc je z genami już znanymi jako markery złośliwości, zaangażowania odpornościowego i przebudowy tkanki, a także wyróżniając dodatkowe, prawdopodobne kandydatury.
Spójne mapy w kolejnych przekrojach tkanki
Prawdziwe tkanki są trójwymiarowe, podczas gdy przestrzenna transkryptomika zwykle mierzy cienkie, dwuwymiarowe przekroje. HarveST potrafi analizować wspólnie kilka kolejnych przekrojów, budując wspólną sieć, która respektuje wewnętrzny układ każdego przekroju, a jednocześnie łączy podobne regiony między przekrojami. W sparowanych sekcjach ludzkiego mózgu ta wspólna analiza poprawiła wyrównanie między przekrojami i wygenerowała ciągłe warstwy korowe, które płynnie rozciągały się z jednej sekcji na drugą. Sugeruje to, że HarveST może pomóc w rekonstrukcji wierniejszych trójwymiarowych widoków architektury tkanki na podstawie standardowych eksperymentów.
Co to oznacza dla przyszłych badań biomedycznych
Podsumowując, HarveST oferuje zunifikowany sposób przejścia od surowych przestrzennych map genów do czytelnych regionów tkankowych i ich kluczowych genów, przy jednoczesnym poszanowaniu zarówno struktury przestrzennej, jak i relacji molekularnych. Dla osób niebędących specjalistami oznacza to, że naukowcy zyskują ostrzejsze „mapy pogodowe” aktywności genów w organach i guzach oraz bardziej wiarygodne listy genów wyjaśniające, dlaczego jeden region różni się od drugiego. W miarę rozwoju technologii przestrzennych podejścia takie jak HarveST mogą stać się kluczowymi narzędziami do zrozumienia, jak zorganizowane są zdrowe tkanki, jak choroby takie jak nowotwór przekształcają tę organizację i gdzie można znaleźć nowe markery diagnostyczne lub cele terapeutyczne.
Cytowanie: Feng, J., Yu, T. & Zhang, Y. HarveST uses a heterogeneous graph learning framework to reveal spatial transcriptomics patterns. Commun Biol 9, 681 (2026). https://doi.org/10.1038/s42003-026-09841-2
Słowa kluczowe: przestrzenna transkryptomika, architektura tkanki, uczenie grafowe, mikrośrodowisko nowotworu, geny markerowe