Clear Sky Science · pl
Opracowanie i ocena skutecznego modelu predykcji rozpuszczalności farmaceutyków w rozpuszczalnikach organicznych z użyciem uczenia maszynowego opartego na eXtreme Gradient Boosting
Dlaczego rozpuszczanie leków ma znaczenie
Gdy tabletka trafia do organizmu, najpierw musi się rozpuścić, by zadziałać. Łatwość, z jaką substancja czynna rozpuszcza się w cieczy, wpływa na sposób wytwarzania leku, jego stabilność i efektywność działania. Mierzenie tej „rozpuszczalności” w wielu różnych cieczach i temperaturach jest czasochłonne i kosztowne. W badaniu sprawdzono, czy nowoczesne narzędzie danych — uczenie maszynowe — może pomóc szybko oszacować, jak dobrze związki o charakterze leku rozpuszczają się w powszechnych rozpuszczalnikach organicznych, korzystając jedynie z niewielkiej ilości łatwo dostępnych informacji.
Wybór odpowiedniego rozpuszczalnika do tworzenia kryształów
W produkcji leków często otrzymuje się kryształy substancji czynnej z rozpuszczalnika organicznego. Rozpuszczalnik nie tylko decyduje o ilości odzyskanego ciała stałego, lecz także wpływa na wielkość i formę kryształów, co z kolei może oddziaływać na właściwości leku. Tradycyjnie chemicy wykonują liczne eksperymenty lub korzystają ze złożonych równań termodynamicznych, aby przewidzieć rozpuszczalność. Te ustalone metody mogą być dokładne, ale często wymagają wielu dopasowywanych parametrów lub szczegółowych danych molekularnych, które nie są dostępne we wczesnym etapie rozwoju. Autorzy tego opracowania zadali więc pytanie, czy starannie zaprojektowany model uczenia maszynowego potrafi uchwycić kluczowe trendy rozpuszczalności, pozostając jednocześnie opartym na prostych, fizycznie znaczących wejściach.
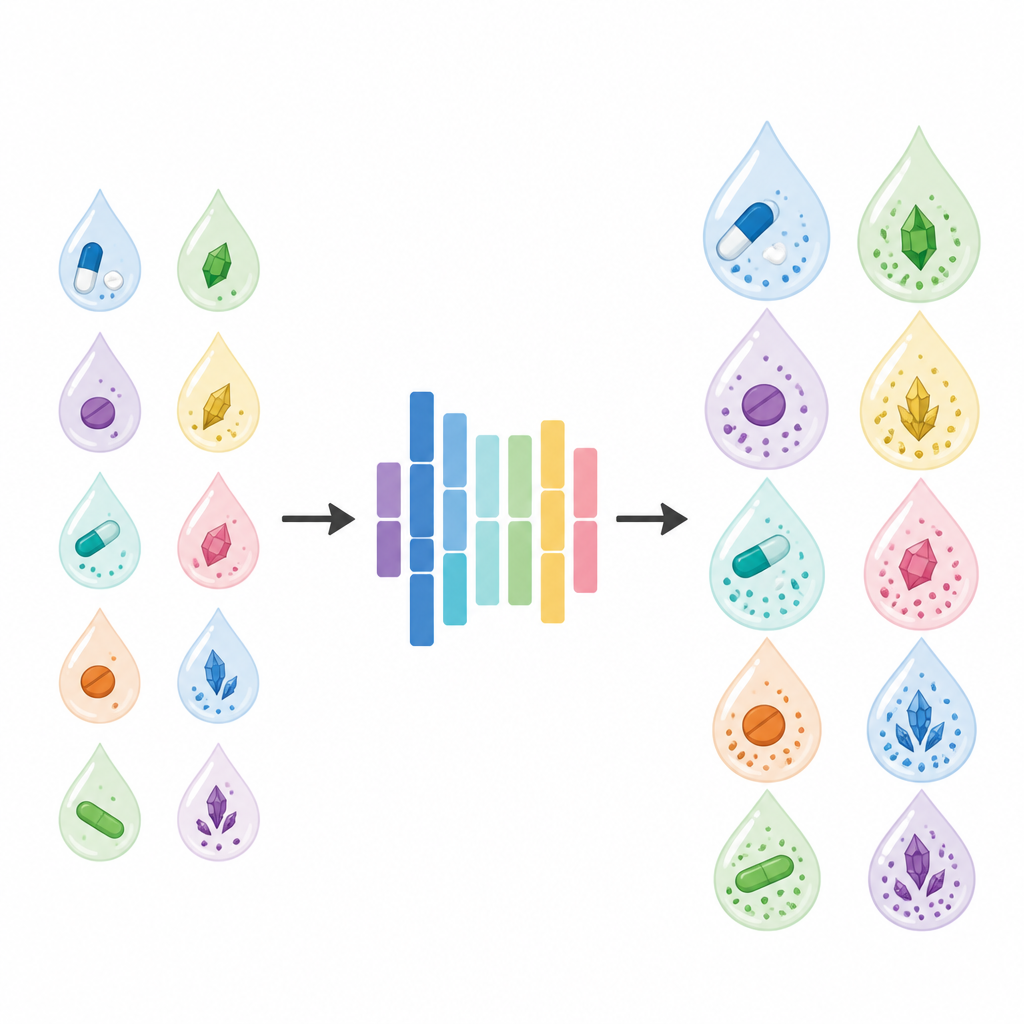
Zwięzły model oparty na danych z fizycznym wglądem
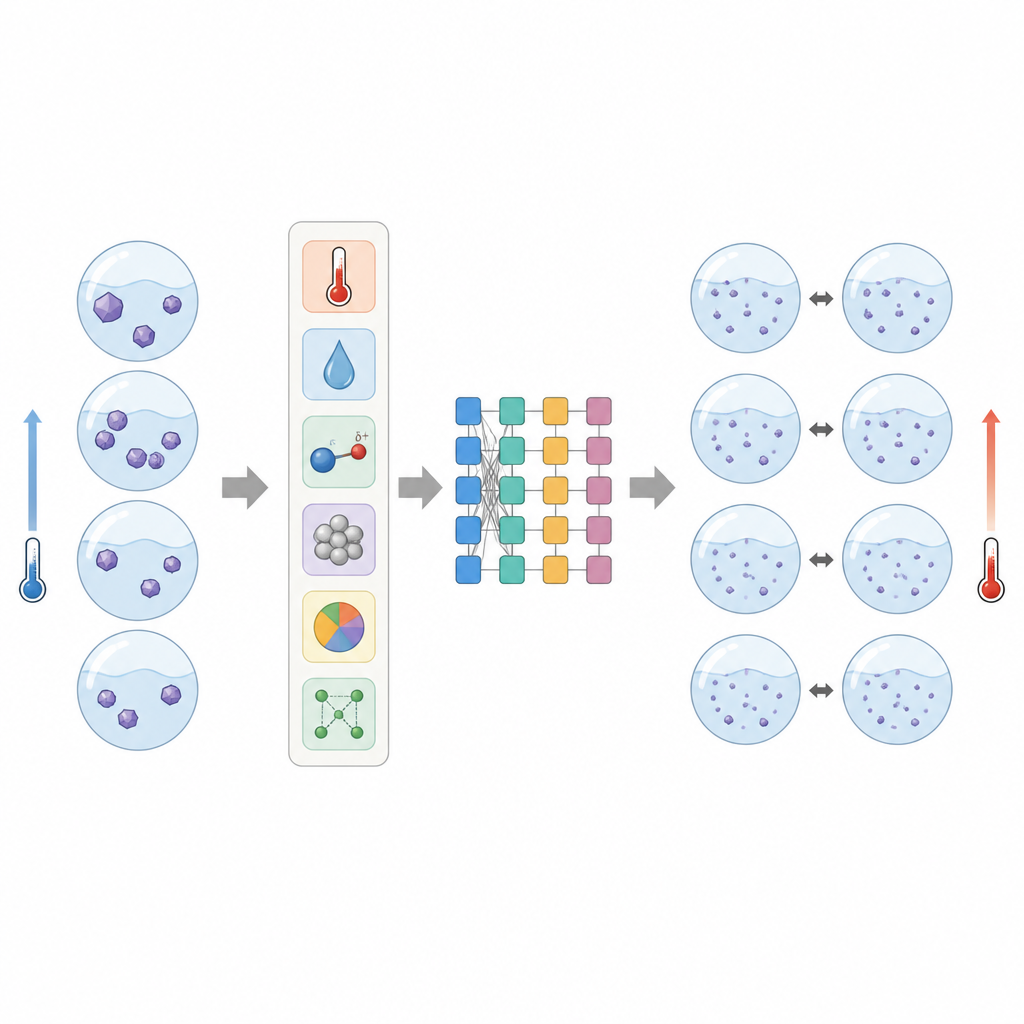
Zespół zbudował model predykcji rozpuszczalności wykorzystując popularną metodę uczenia maszynowego o nazwie eXtreme Gradient Boosting (XGBoost). Zgromadzili opublikowane dane rozpuszczalności dla czterech związków o charakterze leku w dziewięciu powszechnych rozpuszczalnikach organicznych w szerokim zakresie temperatur, otrzymując 224 punktów danych. Zamiast podawać algorytmowi losowe deskryptory, wybrali dziesięć cech zrozumiałych dla chemików: właściwości ciała stałego (takie jak temperatura topnienia, entalpia topnienia, pojemność cieplna oraz dobrze znany parametr rozpuszczalności), podstawowe właściwości cieczy (polaryzacja mierzona stałą dielektryczną i temperatura wrzenia) oraz samą temperaturę i proste kodowania nazw ciała stałego i rozpuszczalnika. Aby odzwierciedlić fakt, że większość substancji rozpuszcza się lepiej po podgrzaniu, wprowadzili regułę zmuszającą przewidywania modelu do wzrostu z temperaturą, co zapewnia fizycznie sensowne zachowanie.
Jak dobrze model zgadza się z rzeczywistymi pomiarami
Po dostrojeniu modelu za pomocą walidacji krzyżowej autorzy sprawdzili, jak bliskie są przewidywania wartościom mierzonym. Oceniali wydajność przez porównanie logarytmów zmierzonej i przewidywanej rozpuszczalności, co jest odpowiednie, ponieważ rozpuszczalności obejmowały kilka rzędów wielkości. Dla czterech związków użytych do treningu i testów model odtworzył dane z bardzo małymi średnimi błędami i wysoką korelacją, co wskazuje, że potrafi wiarygodnie opisać zależność rozpuszczalności od temperatury w różnych środowiskach ciekłych. Co ważne, model pozostał dokładny nawet dla bardzo słabo rozpuszczalnego związku, risperidonu, którego zachowanie jest znane z trudności przy opisie za pomocą prostszych równań.
Przewidywanie całkowicie nowego związku
Kluczowe pytanie brzmiało, czy model poradzi sobie z substancją czynną, której nigdy nie widział. Aby to sprawdzić, badacze odłożyli na bok wszystkie dane dla piątego związku, butambenu, i wykorzystali te 50 pomiarów dopiero po zakończeniu treningu. Błędy modelu były większe dla tego rzeczywistego zadania predykcyjnego niż dla danych, które znał wcześniej, ale nadal mieściły się w zakresie porównywalnym z typową niepewnością eksperymentalną, zwłaszcza dla kilku testowanych rozpuszczalników. W porównaniu z dwoma powszechnie używanymi półpredykcyjnymi metodami termodynamicznymi — Flory-Hugginsa i zależnym od temperatury NRTL-SAC — model XGBoost konsekwentnie generował mniejsze błędy ogółem i wypadał szczególnie dobrze dla najtrudniejszych układów.
Co to oznacza dla przyszłego rozwoju leków
Dla osób niebędących specjalistami kluczowym wnioskiem jest to, że stosunkowo mały, fizycznie ugruntowany model uczenia maszynowego może wiarygodnie oszacować, jak dobrze związki o charakterze leku rozpuszczają się w powszechnych rozpuszczalnikach organicznych w różnych temperaturach. Robi to, korzystając z umiarkowanego zestawu mierzalnych właściwości, bez konieczności intensywnego dopasowywania parametrów typowego dla tradycyjnych podejść. Autorzy zauważają, że dalsze udoskonalenie wybranych deskryptorów i szerszy zbiór danych poprawiłyby wydajność, jednak badanie pokazuje, że takie modele mogą już wspierać dobór rozpuszczalników i projektowanie procesów, pomagając chemikom zawęzić obiecujące opcje przed przeprowadzeniem szczegółowych prac laboratoryjnych.
Cytowanie: Valavi, M., Assareh, M., Khoshsima, A. et al. Development and evaluation of an effective solubility prediction model for pharmaceuticals in organic solvents using machine learning based on eXtreme Gradient Boosting. Sci Rep 16, 16592 (2026). https://doi.org/10.1038/s41598-026-53038-w
Słowa kluczowe: rozpuszczalność leku, rozpuszczalniki organiczne, uczenie maszynowe, XGBoost, krystalizacja