Clear Sky Science · pl
Hydroaminacja Markownikowa terminalnych alkenów za pomocą katalizy redoks fosfinowej
Przekształcanie prostych bloków budulcowych w wartościowe związki azotowe
Wiele leków, środków ochrony roślin i materiałów specjalistycznych opiera się na wiązaniach węgiel–azot, jednak efektywne wprowadzanie azotu do prostych węglowodorowych bloków budulcowych bywa trudne. W artykule opisano nową, napędzaną światłem metodę łączenia powszechnych przemysłowych alkenów z azotowymi pierścieniami, tworząc użyteczne produkty w sposób ściśle kontrolowany. Zastosowanie katalizatora na bazie fosforu zamiast tradycyjnych metali ujawnia reaktywność, której metale często nie osiągają, oferując nową drogę do ważnych związków przy jednoczesnym unikaniu rzadkich lub wrażliwych metali przejściowych.
Dlaczego łączenie węgla z azotem jest tak trudne
Chemicy od dawna poszukują uniwersalnych sposobów dodawania azotu do wiązań podwójnych węgiel–węgiel, reakcji znanej ogólnie jako hydroaminacja. Późne metale przejściowe, takie jak iryd, kobalt czy pallad, katalizują niektóre z tych przemian, ale mają ograniczenia. Przemysłowo obfite, „nieaktywowane” terminalne alkeny — proste łańcuchy jak 1‑heksen — słabo wiążą się z katalizatorami metali i często izomeryzują zamiast reagować czysto. Źródła azotu zwane azolami, powszechne w lekach i agrochemikaliach, mogą również dezaktywować katalizatory metaliczne przez zbyt silne wiązanie lub udział w niepożądanych reakcjach bocznych. W rezultacie nie istniała powszechnie użyteczna metoda oparta na metalach do przeprowadzenia hydroaminacji Markownikowej takich alkenów z szeroką gamą azoli — czyli selektywnego przyłączania azotu do bardziej substytuowanego końca wiązania podwójnego.
Fosfor wchodzi tam, gdzie metale zawodzą
Ostatnie prace wykazały, że pierwiastki grup głównych, takie jak fosfor, mogą naśladować niektóre kluczowe ruchy metali przejściowych, przy jednoczesnej tolerancji innych typów związków. Budując na wcześniejszych badaniach, w których kombinacja fosfiny i fotoredoksu umożliwiła odwrotną selektywność anty‑Markownikową, autorzy odkryli, że zmiana katalizatora fosfinowego odwraca zachowanie reakcji. Z użyciem określonych arylowych fosfin wraz z fotokatalizatorem aktywowanym światłem widzialnym i współkatalizatorem tiolowym osiągnięto hydroaminację terminalnych alkenów z azolami N–H z selektywnością Markownikową. Pod naświetlaniem niebieskim światłem fosfina jest utleniana do wysoce reaktywnego kationu rodnikowego, który aktywuje alken w sposób zwykle przypisywany metalom. Azole mogą następnie atakować ten aktywowany intermediat, tworząc nowe wiązanie węgiel–azot, a kontrolowany etap transferu wodoru kończy produkt i regeneruje katalizatory.
Szeroki wybór pierścieni azotowych i alkenów
Zespół systematycznie zbadał, jak ogólna jest ta metoda. Pokazali, że wiele azoli — pyrazole, imidazole, indazole, benzimidazole, triazole i pokrewne azotowe heterocykle — podlega czystej N‑alkilacji, zwykle w jednym, określonym miejscu azotowym. Nawet złożone, bioaktywne cząsteczki, takie jak neurotransmiter histamina, środek uspokajający dekspmedetomidyna czy antidotum fomepizol, reagują bez zaburzania innych wrażliwych grup funkcyjnych. Po stronie alkenów reaguje szeroka gama terminalnych alkenów alifatycznych, w tym te zawierające estry, acetale, chronione aminy i heterocykle, takie jak pirydyny. Metoda działa na substratach pochodnych naturalnych produktów i steroidów i zwykle wymaga jedynie niewielkiego nadmiaru alkenu — co stanowi przewagę nad wieloma systemami katalizowanymi metalami, które często wymagają dużych nadwyżek surowca. W całym tym szerokim zakresie reakcja dostarcza produktów ze spójną selektywnością Markownikową i wyłączną kontrolą miejsca włączenia azotu.
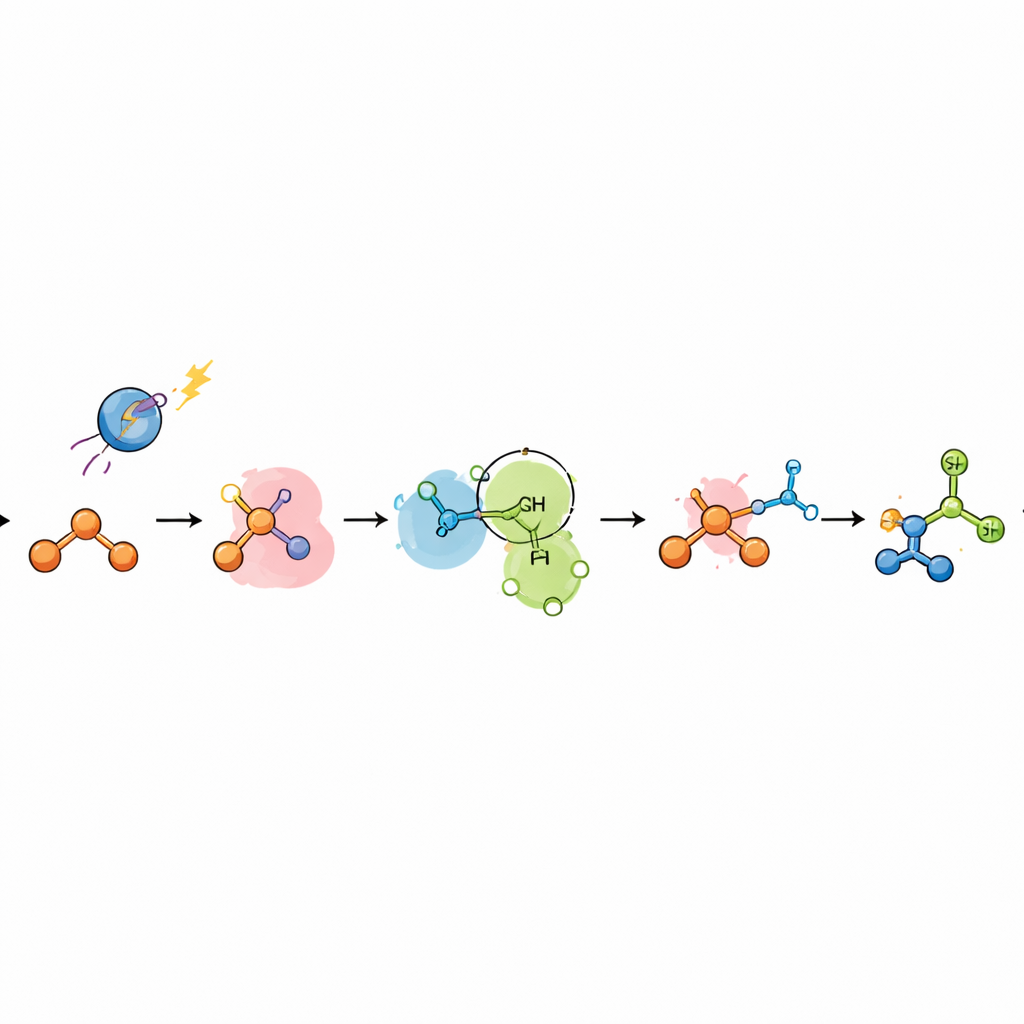
Jak działa nowy trik katalityczny
Aby zrozumieć, dlaczego ten system fosforowy udaje się tam, gdzie metale zawodzą, autorzy połączyli eksperymenty mechanistyczne z obliczeniami kwantowo‑chemicznymi. Badania gaszenia luminescencji wykazały, że to fosfina, a nie azol, jest najpierw utleniana przez wzbudzony fotokatalizator, tworząc kation rodnikowy fosfiny. Ten gatunek addycjonuje do alkenu, generując „distoniczny” kation rodnikowy, w którym ładunek dodatni znajduje się głównie na fosforze, a niesparowany elektron na węglu. Zamiast prostego transferu wodoru, ten intermediat jest atakowany przez azol w kroku polarnym przypominającym nukleofilową addycję metalu do skoordynowanego alkenu. Analizy obliczeniowe i badania kinetyczne wspierają dwie blisko spokrewnione ścieżki: krokową drogę przez pentawalentny intermediat fosforu i wstawienie migrujące oraz drogę koncertowaną, w której atak azolu i wewnętrzny transfer elektronów zachodzą jednocześnie, podobnie do mikroskopowego odwrotu znanego procesu rodnikowego „przesunięcia centrum spinowego”. W obu przypadkach radikal fosforylowy ostatecznie rozrywa swoje wiązanie węgiel–fosfor, generując rodnik węglowy w pozycji Markownikowej, który następnie pobiera wodór od współkatalizatora tiolowego.
Pisanie od nowa zasad dla katalizatorów nie‑metalicznych
Pokazując, że kation rodnikowy fosforu może pośredniczyć w kluczowym kroku zwykle zarezerwowanym dla metali przejściowych — nukleofilowej funkcjonalizacji alkenu z selektywnością Markownikową — praca ta poszerza przestrzeń projektową dla syntezy katalitycznej. Dostarcza praktycznej metody konstrukcji wiązań węgiel–azot z łatwo dostępnych alkenów i farmaceutycznie istotnych azoli w łagodnych, redoks‑neutralnych warunkach, z szeroką tolerancją grup funkcyjnych. Szerzej, wgląd mechanistyczny w to, jak distoniczne rodniki fosforu angażują nukleofile, sugeruje rodzinę jeszcze nieodkrytych reakcji, w których pierwiastki grup głównych mogą rywalizować z metalami przejściowymi lub nawet je przewyższać w umożliwianiu złożonej budowy molekularnej.
Cytowanie: Fan, F., Sedillo, K.F., Maertens, A.J. et al. Markovnikov hydroamination of terminal alkenes by phosphine redox catalysis. Nature 652, 96–104 (2026). https://doi.org/10.1038/s41586-026-10263-7
Słowa kluczowe: hydroaminacja, kataliza fosfinowa, chemia fotoredoks, funkcjonalizacja alkenów, N‑alkilacja azoli