Clear Sky Science · en
Markovnikov hydroamination of terminal alkenes by phosphine redox catalysis
Turning Simple Building Blocks into Valuable Nitrogen Compounds
Many medicines, crop-protection agents and specialty materials rely on carbon–nitrogen bonds, but efficiently stitching nitrogen onto simple hydrocarbon building blocks is often tricky. This paper describes a new light-driven method that joins common industrial alkenes with nitrogen‑rich ring molecules to form useful products in a highly controlled way. By using a phosphorus-based catalyst instead of traditional metals, the researchers unlock reactivity that metals struggle to achieve, offering a fresh route to important molecules while avoiding scarce or sensitive transition metals.
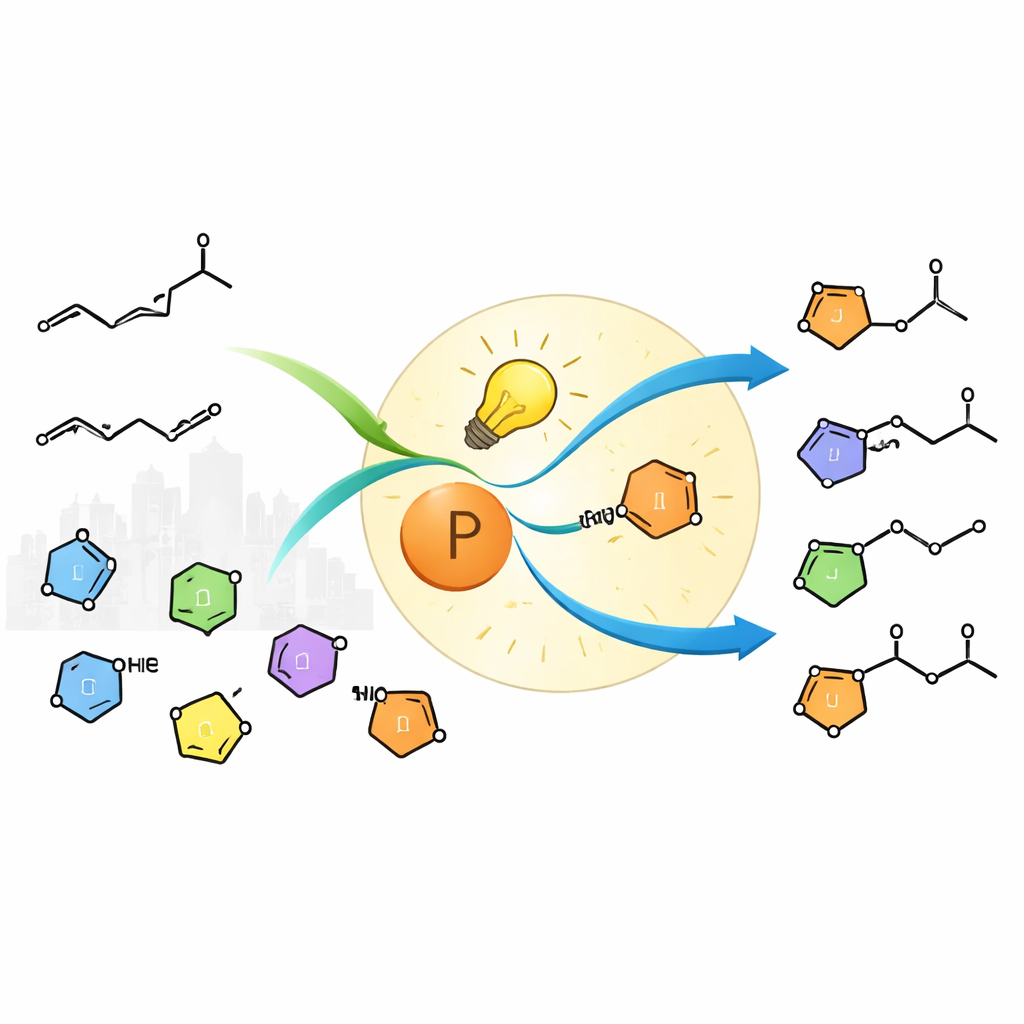
Why Joining Carbon and Nitrogen Is So Challenging
Chemists have long sought general ways to add nitrogen across carbon–carbon double bonds, a transformation known broadly as hydroamination. Late transition metals such as iridium, cobalt and palladium can catalyze some of these reactions, but they have blind spots. Industrially abundant "unactivated" terminal alkenes—simple chains like 1‑hexene—bind weakly to metal catalysts and often isomerize instead of reacting cleanly. Nitrogen sources called azoles, which are common in drugs and agrochemicals, can also shut down metal catalysts by clinging too tightly or engaging in unwanted side reactions. As a result, there has been no broadly useful metal-based method to perform Markovnikov hydroamination of such alkenes with a wide variety of azoles—that is, selectively attaching nitrogen to the more substituted end of the double bond.
Phosphorus Steps In Where Metals Struggle
Recent work has shown that main‑group elements like phosphorus can mimic some of the key moves of transition metals while tolerating different types of molecules. Building on earlier studies where a phosphine–photoredox combination enabled the opposite, anti‑Markovnikov selectivity, the authors discovered that changing the phosphine catalyst flips the reaction’s behavior. Using specific aryl phosphines together with a visible‑light photocatalyst and a thiol co‑catalyst, they achieved Markovnikov‑selective hydroamination of terminal alkenes with N–H azoles. Under blue‑light irradiation, the phosphine is oxidized to a highly reactive radical cation that activates the alkene in a way normally associated with metals. Azoles can then attack this activated intermediate to forge a new carbon–nitrogen bond, and a controlled hydrogen‑transfer step finishes the product while regenerating the catalysts.
A Broad Menu of Nitrogen Rings and Alkenes
The team systematically explored how general this process is. They showed that many azoles—pyrazoles, imidazoles, indazoles, benzimidazoles, triazoles, and related nitrogen heterocycles—undergo clean N‑alkylation, typically at a single nitrogen site. Even complex, bioactive molecules such as the neurotransmitter histamine, the sedative dexmedetomidine, and the antidote fomepizole participate without disturbing other sensitive functional groups. On the alkene side, a wide range of terminal aliphatic alkenes, including those bearing esters, acetals, protected amines, and heterocycles like pyridines, react efficiently. The method works with substrates derived from natural products and steroids, and usually requires only a modest excess of the alkene—an advantage over many metal‑catalyzed systems, which often demand large surpluses of feedstock. Across this broad scope, the reaction delivers products with consistent Markovnikov selectivity and exclusive nitrogen‑site control.
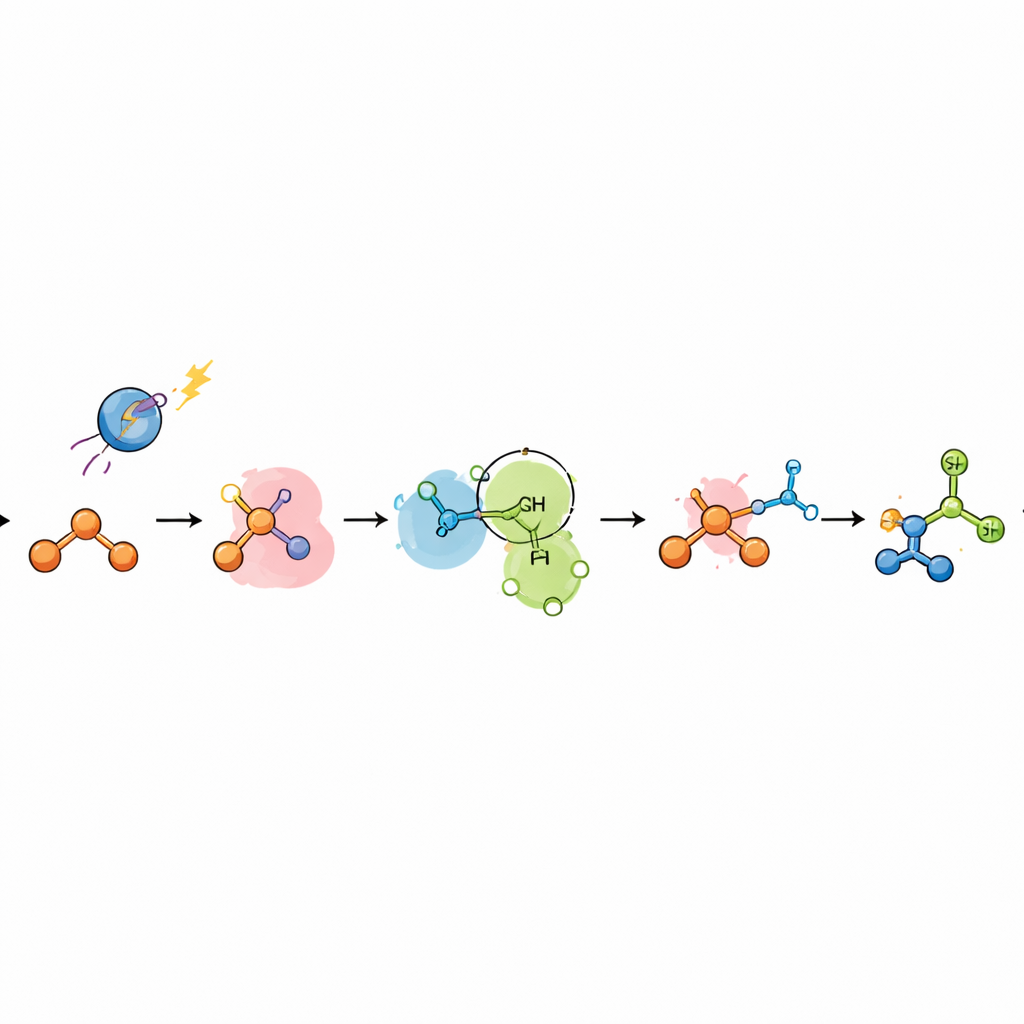
How the New Catalytic Trick Works
To understand why this phosphorus system succeeds where metals falter, the authors combined mechanistic experiments with quantum‑chemical calculations. Luminescence quenching studies showed that the phosphine, not the azole, is first oxidized by the excited photocatalyst, forming a phosphine radical cation. This species adds to the alkene to generate a "distonic" radical cation, in which positive charge resides mainly on phosphorus and the unpaired electron on carbon. Rather than undergoing simple hydrogen transfer, this intermediate is attacked by the azole in a polar step that resembles metal‑mediated nucleophilic addition to a coordinated alkene. Computational analyses and kinetic probes support two closely related pathways: a stepwise route via a pentavalent phosphorus intermediate and migratory insertion, and a concerted route in which azole attack and internal electron transfer occur together, akin to the microscopic reverse of a known radical "spin‑center shift" process. In both cases, a phosphoranyl radical ultimately breaks its carbon–phosphorus bond to generate a carbon‑centered radical at the Markovnikov position, which then picks up hydrogen from the thiol co‑catalyst.
Rewriting the Rules for Non‑Metal Catalysts
By showing that a phosphorus radical cation can mediate a key step normally reserved for transition metals—nucleophilic functionalization of an alkene with Markovnikov selectivity—this work broadens the design space for catalytic synthesis. It delivers a practical method for constructing carbon–nitrogen bonds from readily available alkenes and medicinally important azoles under mild, redox‑neutral conditions, with wide functional‑group tolerance. More broadly, the mechanistic insights into how distonic phosphorus radicals engage nucleophiles hint at a family of yet‑to‑be‑developed reactions in which main‑group elements rival, or even surpass, transition metals in enabling complex molecular construction.
Citation: Fan, F., Sedillo, K.F., Maertens, A.J. et al. Markovnikov hydroamination of terminal alkenes by phosphine redox catalysis. Nature 652, 96–104 (2026). https://doi.org/10.1038/s41586-026-10263-7
Keywords: hydroamination, phosphine catalysis, photoredox chemistry, alkene functionalization, azole N-alkylation