Clear Sky Science · pl
Wykrywanie transdukcji bakteriofagów między rzędami w społecznościach mikrobiologicznych za pomocą znakowania RNA
Wirusy, które cicho przepisują życie mikrobiologiczne
Niewidoczne armie wirusów nieustannie wymieniają geny między bakteriami w naszych jelitach, ciekach wodnych i glebach, przekształcając ekosystemy i wpływając na zdrowie człowieka. Naukowcy jednak mieli trudności z ustaleniem, który wirus zakaża konkretny mikroorganizm, zwłaszcza w złożonych, rzeczywistych społecznościach, takich jak ścieki. W tym badaniu wprowadzono sprytny molekularny notatnik zintegrowany z wirusem, który pozwala badaczom zapisać, kto kogo zainfekował, ujawniając ukryte powiązania, które mogą pomóc w bezpieczniejszym stosowaniu terapii przeciwdrobnoustrojowych i w inżynierii mikrobiomów.
Dlaczego śledzenie małych kurierów genów ma znaczenie
Bakteriofagi, czyli fagi, to wirusy zakażające bakterie. Mogą zabijać swoje gospodarz lub dyskretnie dostarczać nowe geny, w tym geny oporności na antybiotyki lub korzystne cechy metaboliczne. Przy szacowanej liczbie 1031 cząstek wirusowych na Ziemi są kluczowymi graczami w ewolucji i funkcjonowaniu społeczności mikrobiologicznych. Fagi są także badane jako celowane alternatywy dla antybiotyków oraz jako nośniki narzędzi genetycznych. Aby korzystać z nich rozsądnie, naukowcy muszą wiedzieć, które bakterie każdy fag może zakażać w złożonych społecznościach, a nie tylko w czystych hodowlach laboratoryjnych.
Ograniczenia starszych metod detekcji
Tradycyjne podejścia do mapowania par fag–gospodarz opierają się w dużej mierze na testach płytkowych (plaque assays), które działają tylko wtedy, gdy zarówno wirus, jak i gospodarz mogą być hodowani i testowani pojedynczo w laboratorium. Inne metody mogą wykazać, które fagi przylegają do powierzchni bakterii lub łączyć wirusowe DNA z genomami bakteryjnymi, ale często wymagają rozległej obróbki próbek, specjalistycznych urządzeń lub kosztownego sekwencjonowania całych społeczności. Wiele z tych narzędzi ma też problem z odróżnieniem wirusa, który jedynie dotknął komórki, od tego, który skutecznie wprowadził do niej DNA — a to jest kluczowy krok dla transferu genów.
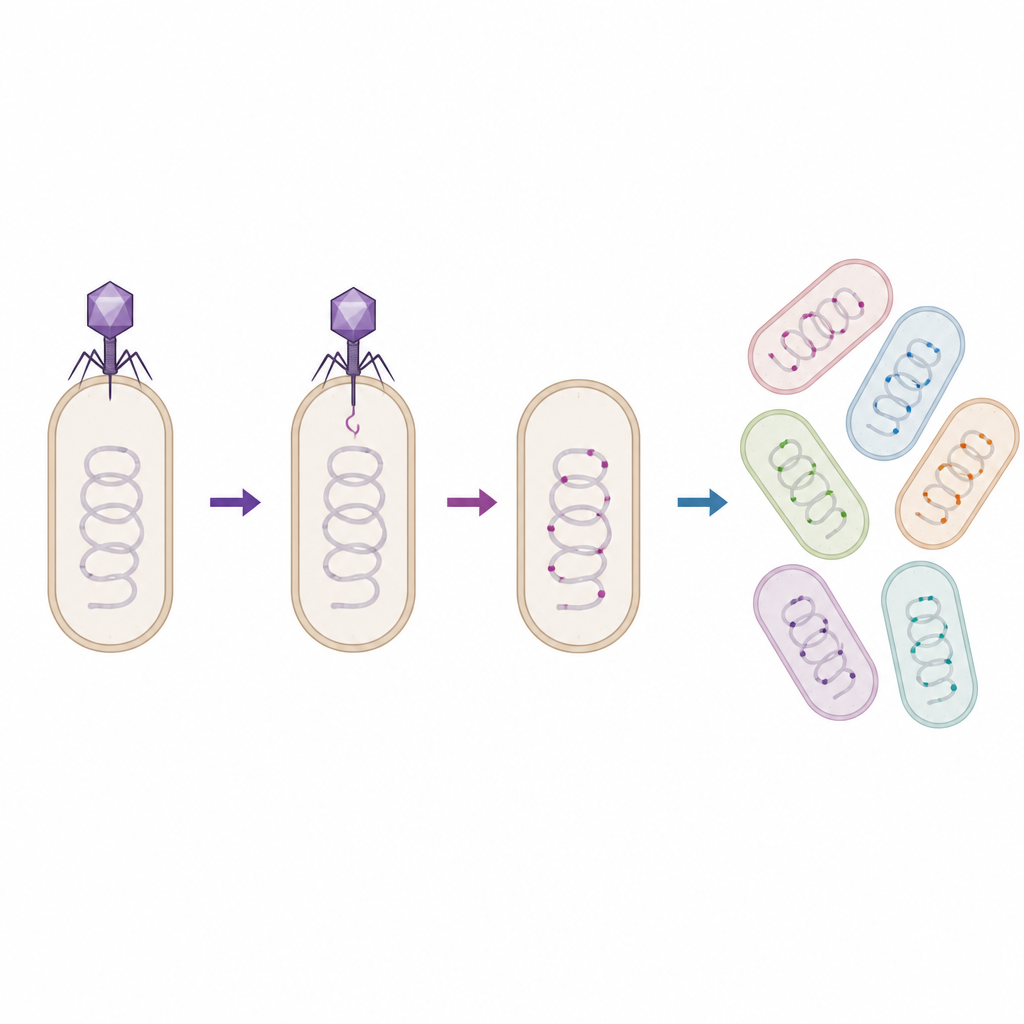
Molekularny kod kreskowy zapisany w bakteryjnym RNA
Zespół zaadaptował narzędzie inżynierii syntetycznej zwane modyfikacją dostępną przez RNA, w skrócie RAM, aby rozwiązać ten problem. Zmodyfikowali powszechny fag o nazwie P1 oraz powiązane z nim cząstki pochodzące od fagów zwane phagemidami, aby przenosiły małą maszynę RNA znaną jako rybozym. Gdy zaprojektowany wirus pomyślnie wejdzie do komórki bakteryjnej, rybozym przyłącza krótki sztuczny „kod kreskowy” do 16S rybosomalnego RNA gospodarza — cząsteczki obecnej we wszystkich bakteriach i powszechnie używanej do identyfikacji gatunków. Później badacze mogą selektywnie sekwencjonować te oznakowane fragmenty RNA, aby odczytać, które członki społeczności zostały rzeczywiście transdukowane, korzystając ze standardowych procedur laboratoryjnych.
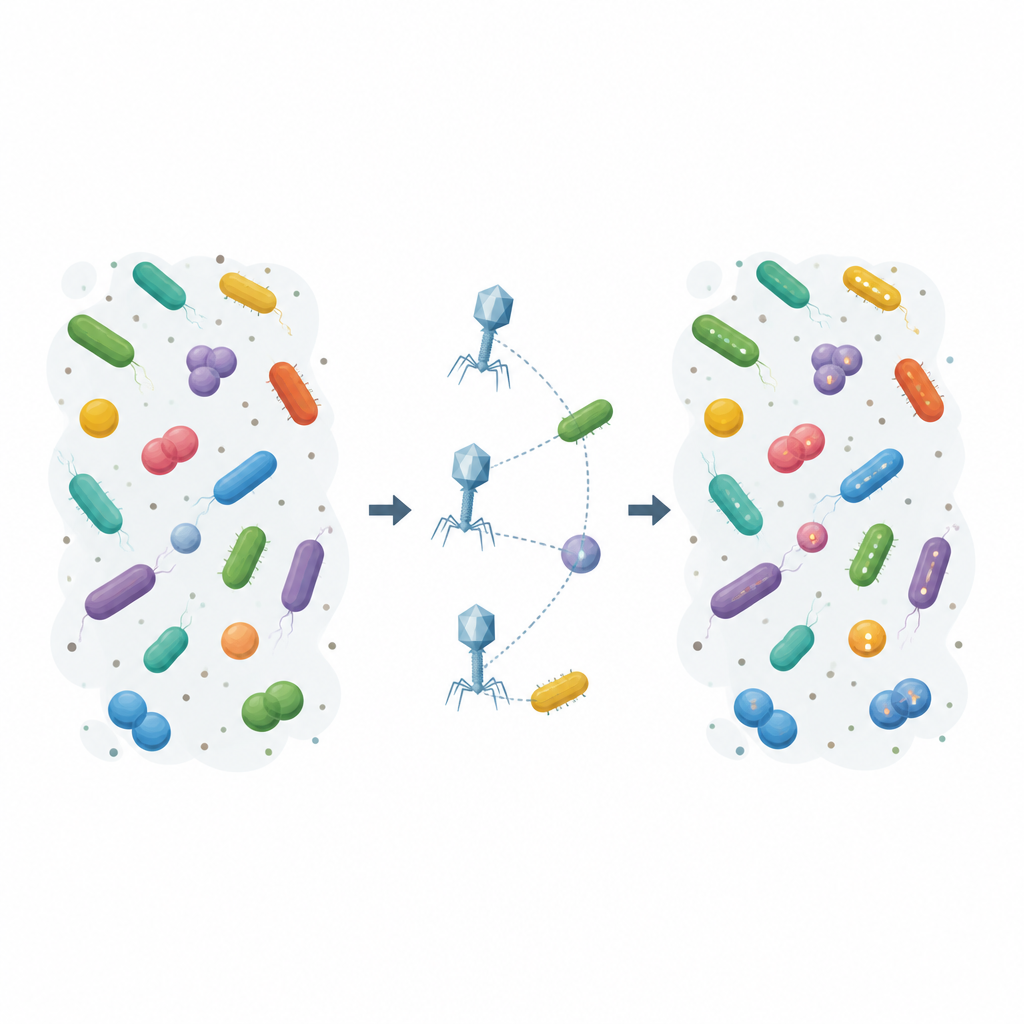
Ujawnianie ukrytych gospodarzy w społecznościach laboratoryjnych i ściekowych
Najpierw autorzy pokazali, że P1 i phagemidy niosące RAM mogą niezawodnie znakować zakażone komórki u kilku dobrze poznanych bakterii jelitowych, a siła sygnału kodu kreskowego odzwierciedlała różnice w efektywności rozprzestrzeniania się każdego konstruktu. Następnie przeszli do syntetycznej ośmiogatunkowej społeczności zawierającej ważne dla człowieka patogeny i ich krewnych. W tym układzie system RAM zarejestrował, które bakterie otrzymały wirusowe DNA, odkrywając nowe zdarzenia transdukcji, w tym stabilne dostarczenie phagemidu o szerokim zakresie gospodarzy do Salmonella enterica. Ponieważ kod kreskowy jest zapisywany w RNA produkowanym przez gospodarza, metoda mogła wykrywać zakażenia także wtedy, gdy zwykłe sztuczki z selekcją antybiotykową były niewykonalne.
Odkrycia w rzeczywistym, mikrobiologicznym rosole
Następnie badacze zastosowali znakowane cząstki P1 do próbek napływowych ścieków bogatych w różnorodne mikroby. Sekwencjonowanie znakowanego RNA ujawniło, że mniej więcej połowa wykrywalnych wariantów sekwencji bakteryjnych w tym środowisku otrzymała wirusowe DNA od przynajmniej jednego z konstrukcji. Co uderzające, metoda wskazała Aeromonas, powszechny rodzaj występujący w ściekach, jako dotychczas nierozpoznanego gospodarza dla P1. Dalsze eksperymenty z wyizolowanymi szczepami Aeromonas potwierdziły, że przynajmniej jeden gatunek mógł być transdukowany i produkował znakowane RNA, demonstrując, jak ta strategia może ujawniać nowe powiązania wirus–gospodarz, których standardowe hodowle by nie wykryły.
Jak budowa ogonka wirusa przekształca mapę zakażeń
Ponad katalogowaniem gospodarzy, zespół użył systemu RAM do zbadania, co kontroluje, które bakterie P1 może zakażać. Skupili się na dwóch naturalnie przełączalnych włóknach ogonkowych wirusa, które rozpoznają różne struktury cukrowe na powierzchniach bakterii. Konstruując cząstki niosące jeden lub drugi typ ogonka i śledząc znakowane RNA w społecznościach ściekowych, wykazali, że te alternatywne ogonki generowały odmienne profile zakażeń. Na przykład cząstki z ogonkiem S′ preferowały niektóre rodzaje związane z jelitami, takie jak Enterobacter i Klebsiella, podczas gdy mieszanki zawierające oba ogonki docierały do jeszcze szerszego zestawu celów, w tym Aeromonas i Acinetobacter.
Co to oznacza dla przyszłych narzędzi fagowych
Łącznie te eksperymenty ustanawiają znakowanie RNA jako elastyczny, skalowalny sposób odczytywania, kogo fagi zakażają w złożonych, niewyhodowanych społecznościach. Metoda opiera się na krótkim sekwencjonowaniu celowanym zamiast pełnych metagenomów, co obniża koszty przy zachowaniu zdolności przypisywania gospodarzy na poziomie mniej więcej gatunku lub rodzaju. Chociaż nie rozróżnia jeszcze bardzo blisko spokrewnionych szczepów ani nie gwarantuje, że wirus ukończył cały cykl życiowy, oferuje praktyczny schemat do przesiewania dużych paneli inżynierowanych fagów lub projektów włókien ogonkowych. W dłuższej perspektywie takie znakowane fagi mogą pomóc badaczom precyzyjniej dopasować terapie wirusowe do problematycznych bakterii i zrozumieć, jak przemieszczanie się genów za pośrednictwem wirusów kształtuje zdrowie i stabilność mikrobiomów.
Cytowanie: LaTurner, Z.W., Dysart, M.J., Schwartz, S.K. et al. Cross-order detection of bacteriophage transduction in microbial communities using RNA barcoding. Nat Commun 17, 4308 (2026). https://doi.org/10.1038/s41467-026-70995-y
Słowa kluczowe: bakteriofag, mikrobiom, znakowanie RNA, poziomy transfer genów, bakterie z ścieków