Clear Sky Science · pl
Wysokorozdzielcze przypisanie fazów do gospodarzy za pomocą kluczowych białek i dużych modeli językowych
Polowanie na niewidoczne wirusy w naszych jelitach
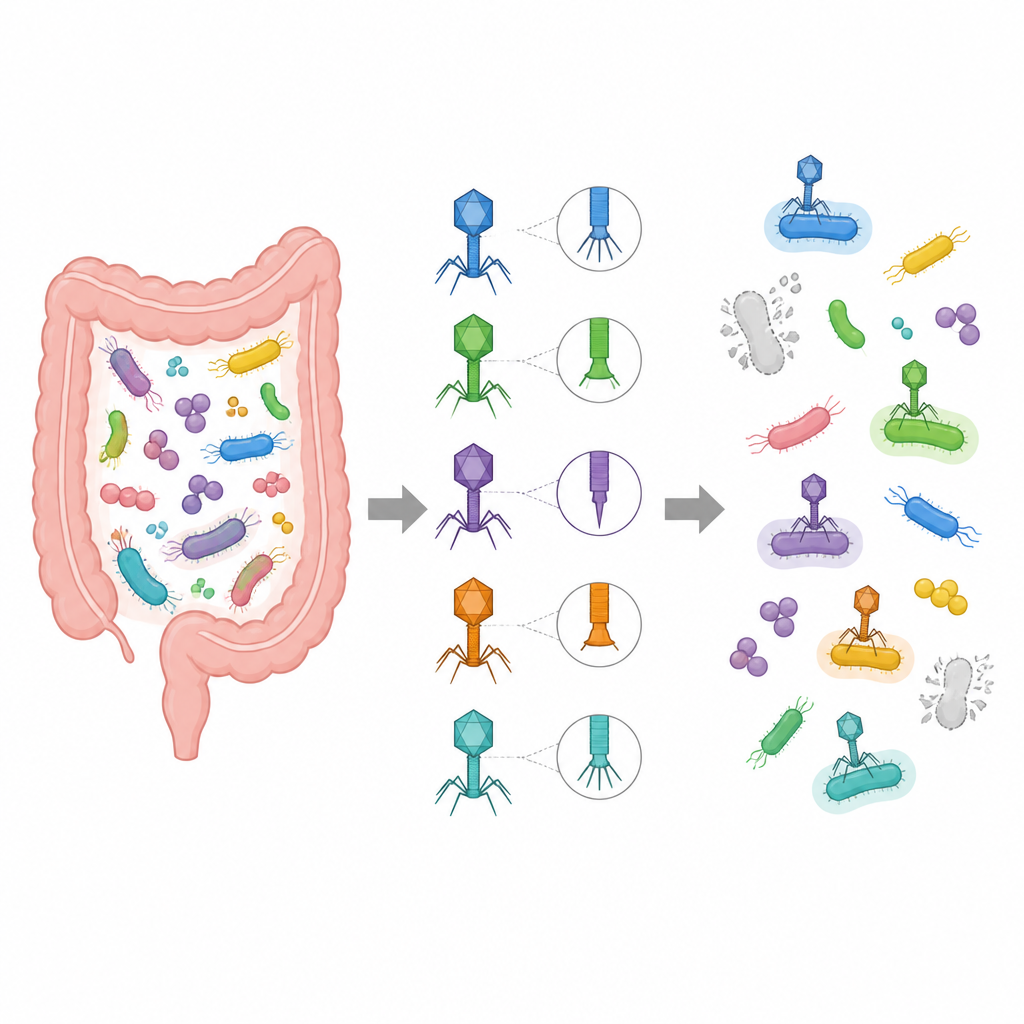
Każdy z nas nosi w jelitach biliony bakterii i ich wirusów, z których wiele wciąż pozostaje nieznanych. Te ukryte wirusy mogą wpływać na nasze zdrowie — od trawienia po otyłość — jednak naukowcy często nie wiedzą, który wirus infekuje którą bakterię. W tym badaniu autorzy przedstawiają VirHost Hunter, nowe narzędzie oparte na danych, które łączy wirusy jelitowe z ich bakteryjnymi gospodarzami, korzystając jedynie z kilku kluczowych białek wirusowych, otwierając drogę do precyzyjniejszych badań i potencjalnej kontroli mikrobiomu.
Nowy sposób dopasowywania wirusów do bakterii
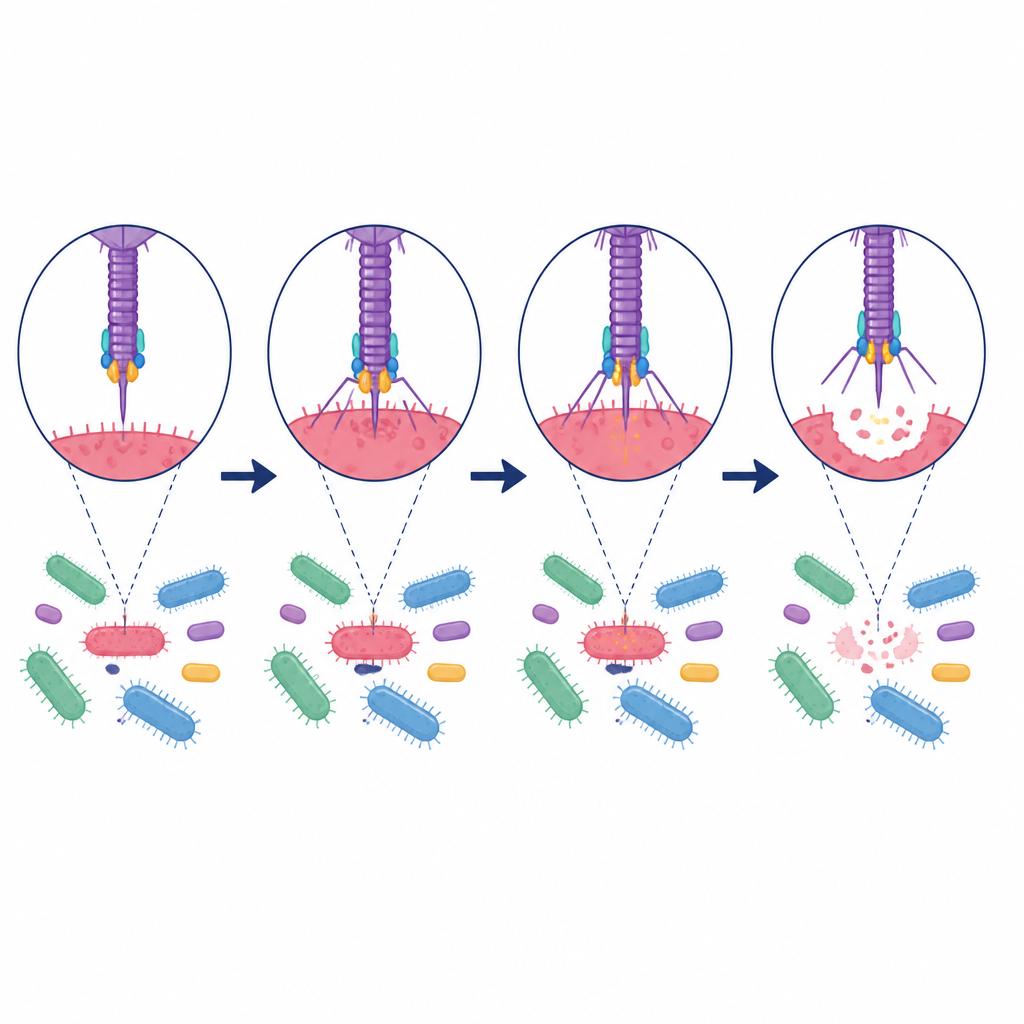
Tradycyjne metody parowania wirusów z ich gospodarzami bazują na pełnych genomach wirusowych lub specjalnych wskazówkach genetycznych, takich jak markery CRISPR. Podejścia te działają tylko wtedy, gdy istnieją odpowiednie dane referencyjne i mogą pominąć dużą część sekwencji wirusowych, często określanych jako wirusowa ciemna materia. Autorzy skupiają się zamiast tego na dwóch typach białek wirusowych kluczowych dla infekcji: białkach ogonowych, które pomagają wirusowi rozpoznać i przyczepić się do bakterii, oraz lizynach, które pozwalają mu rozbić ścianę komórkową bakterii. Koncentrując się na tych białkach, unikają szumu pochodzącego od niepowiązanych genów i mogą działać nawet wtedy, gdy dostępne są tylko fragmenty genomu wirusowego.
Nauczanie komputerów języka białek i DNA
Aby wydobyć znaczenie z tych białek, zespół sięga po techniki uczenia maszynowego pierwotnie opracowane do analizy języka ludzkiego. Wykorzystują model językowy białek o nazwie ProtT5, który przekształca sekwencje aminokwasów w gęste reprezentacje numeryczne uchwytujące ukryte podobieństwa funkcjonalne, nawet gdy sekwencje początkowo wyglądają bardzo różnie. Równolegle analizują DNA kodujące te białka za pomocą modelu Vision Transformer i wielościeżkowej sieci konwolucyjnej, które razem wychwytują cechy takie jak typowe wykorzystanie kodonów i długozasięgowe wzorce wzdłuż DNA. Sygnały z białek i DNA są następnie scalane i podawane do pary klasyfikatorów, które wspólnie decydują, jakiej rodziny, rodzaju czy gatunku bakteryjnego dany wirus najprawdopodobniej infekuje.
Bardziej precyzyjne i głębsze prognozy gospodarzy
Badacze przetestowali VirHost Hunter na kilku zestawach referencyjnych bakteriofagów. Pokazują, że łączenie informacji z białek i DNA wyraźnie przewyższa użycie jednego z tych źródeł osobno, a koncentracja na białkach ogonowych i lizynach daje lepsze przewidywania niż wykorzystywanie innych fragmentów wirusa, takich jak kapsydy czy enzymy pakujące. Na różnych poziomach klasyfikacji bakteryjnej VirHost Hunter jest dokładniejszy niż istniejące narzędzia wolne od wyrównywania i pozostaje wiarygodny nawet wtedy, gdy wirusy wykazują niskie podobieństwo sekwencyjne. W ocenie na hodowanych faga jelitowych o eksperymentalnie znanych gospodarzach identyfikuje poprawnych gospodarzy z wyższą precyzją niż standardowa metoda oparta na CRISPR, a połączenie obu podejść dodatkowo poprawia wyniki.
Ujawnianie ukrytych wirusów jelitowych związanych z chorobami
Wyposażeni w skalibrowany model, autorzy zastosowali VirHost Hunter do dużej bazy Gut Phage Database, która wcześniej miała informacje o gospodarzu dla mniej niż jednej trzeciej wpisów. Skanując białka ogonowe i lizyny, niemal podwoili udział fagów z przypisanymi gospodarzami i odkryli wirusy atakujące 29 rodzin bakterii jelitowych, wiele powiązanych z przewlekłymi schorzeniami, takimi jak choroby zapalne jelit, choroby serca i otyłość. Co istotne, znaleźli dziesiątki wcześniej nieopisanych fagów przewidywanych do infekowania bakterii takich jak Akkermansia muciniphila i Prevotella copri, które były powiązane z chorobami autoimmunologicznymi i metabolicznymi, lecz pozbawione znanych fagów.
Od cyfrowych prognoz do ukierunkowanego środka przeciwdrobnoustrojowego
Aby przekształcić te przewidywania w praktyczne zasoby, autorzy zbudowali Gut Phage Lysin Database zawierającą ponad sto tysięcy lizyn z przypisanymi gospodarzami jelitowymi. Zbadali ich struktury, stabilność i różnorodność, ujawniając wiele odrębnych klastrów i zachowanych motywów odpowiedzialnych za rozrywanie ścian komórkowych bakterii. Jako dowód koncepcji wybrali jedną lizynę przewidywaną do specyficznego zaatakowania Megamonas — bakterii powiązanej z otyłością. Po zsyntezowaniu tego białka wykazali w testach laboratoryjnych, że skutecznie zabija Megamonas, oszczędzając przy tym inne powszechne mikroby jelitowe i szczepy probiotyczne, co ilustruje, jak sterowane modelem eksplorowanie wirusowej ciemnej materii może dostarczyć wysoce selektywnych narzędzi.
Dlaczego to ma znaczenie dla przyszłej opieki nad mikrobiomem
To badanie pokazuje, że możliwe jest powiązanie ogromnych ilości nieznanych wirusów jelitowych z ich bakteryjnymi gospodarzami, używając jedynie kilku kluczowych białek i współczesnego uczenia maszynowego. Oświecając, kto kogo infekuje w mikrobiomie, VirHost Hunter zwiększa naszą zdolność do badania różnorodności wirusów jelitowych i projektowania precyzyjnych interwencji, takich jak dopasowane lizyny, które selektywnie ograniczają szkodliwe bakterie bez zaburzania szerszej społeczności mikrobiologicznej. Choć przed zastosowaniem klinicznym potrzebne są dalsze testy i inżynieria, ramy te dostarczają silnej mapy drogowej do przekształcania ukrytych sekwencji wirusowych w ukierunkowane strategie badawcze i, pewnego dnia, regulacyjne narzędzia do kształtowania naszego wewnętrznego ekosystemu.
Cytowanie: Du, Z., Li, M., Lin, K. et al. High-resolution phage-host assignment through key proteins using large language models. Nat Commun 17, 4439 (2026). https://doi.org/10.1038/s41467-026-70613-x
Słowa kluczowe: wirion jelitowy, bakteriofagi, uczenie maszynowe, lizyny fazowe, terapia mikrobiomu