Clear Sky Science · it
Assegnazione ad alta risoluzione fago-ospite tramite proteine chiave usando modelli di linguaggio di grandi dimensioni
Caccia ai virus invisibili nel nostro intestino
Ogni persona ospita trilioni di batteri e dei loro virus nell’intestino, molti dei quali sono ancora sconosciuti. Questi virus nascosti possono influenzare la nostra salute, dalla digestione all’obesità, eppure gli scienziati spesso non sanno quale virus infetti quale batterio. Questo studio introduce VirHost Hunter, un nuovo strumento guidato dai dati che collega i virus intestinali ai loro batteri ospiti usando solo poche proteine virali chiave, aprendo la strada a modi più precisi per studiare e potenzialmente orientare il microbioma.
Un nuovo modo di abbinare virus e batteri
I metodi tradizionali per associare i virus ai loro batteri ospiti si basano su genomi virali completi o su indizi genetici speciali come i markers CRISPR. Questi approcci funzionano solo quando esistono dati di riferimento adeguati e possono perdere una gran parte delle sequenze virali, spesso chiamate materia oscura virale. Gli autori si concentrano invece su due tipi di proteine virali centrali per l’infezione: le proteine della coda, che aiutano il virus a riconoscere e agganciarsi a un batterio, e le lisine, che gli consentono di rompere la parete cellulare batterica. Concentrandosi su queste proteine evitano il rumore di geni non correlati e possono lavorare anche quando sono disponibili solo frammenti di un genoma virale.
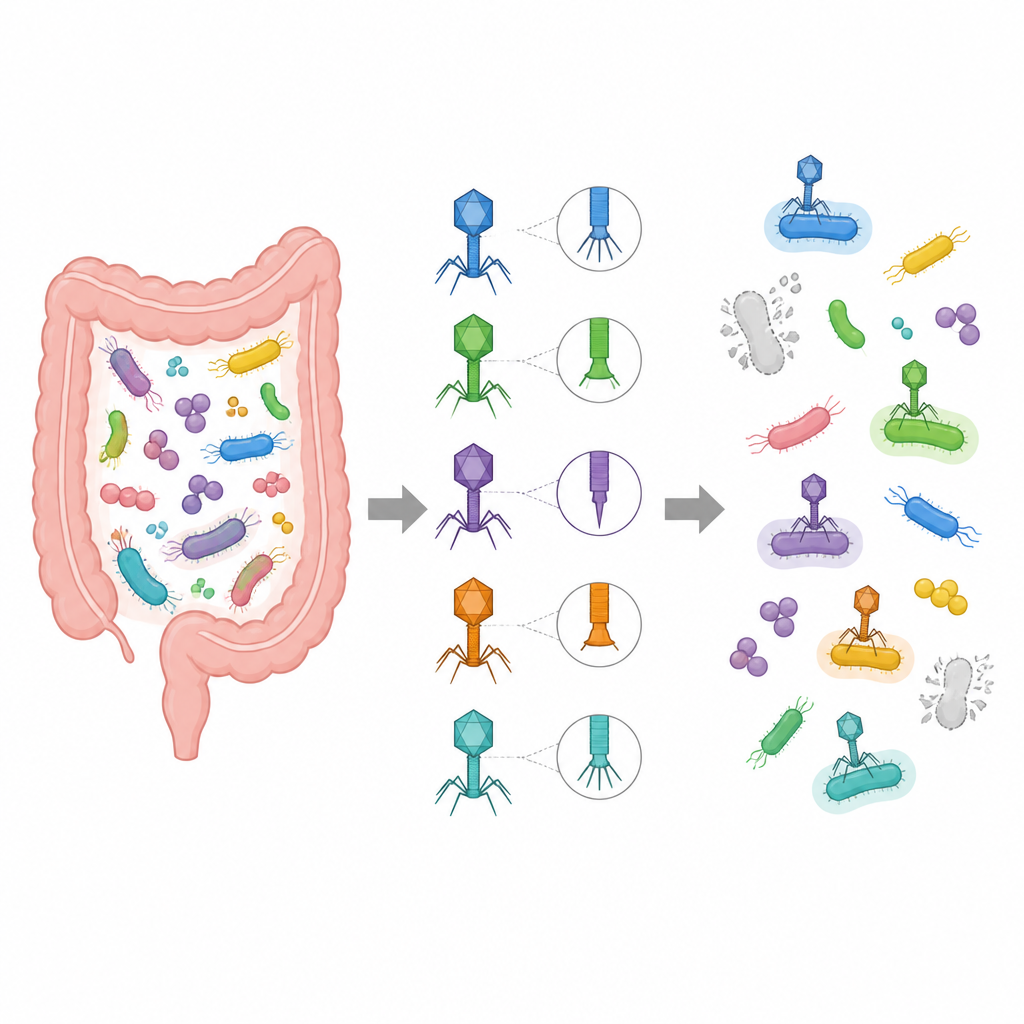
Insegnare ai computer il linguaggio delle proteine e del DNA
Per leggere il significato in queste proteine, il team ricorre a tecniche di apprendimento automatico sviluppate originariamente per il linguaggio umano. Usano un modello linguistico per proteine chiamato ProtT5 per trasformare sequenze di amminoacidi in schemi numerici densi che catturano somiglianze funzionali nascoste, anche quando le sequenze sembrano molto diverse a prima vista. Parallelamente, analizzano il DNA che codifica queste proteine usando un modello Vision Transformer e una rete convoluzionale multi-percorso, che insieme rilevano caratteristiche come l’uso tipico dei codoni e pattern a lungo raggio lungo il DNA. Questi segnali proteici e del DNA vengono poi fusi e immessi in una coppia di classificatori che decidono congiuntamente quale famiglia, genere o specie batterica un dato virus è probabile che infetti.
Predizioni dell’ospite più nitide e profonde
I ricercatori hanno testato VirHost Hunter su diverse collezioni di riferimento di batteriofagi. Dimostrano che combinare informazioni proteiche e del DNA supera nettamente l’uso di una sola sorgente, e che concentrarsi su proteine della coda e lisine fornisce previsioni migliori rispetto all’uso di altre parti virali come i capside o gli enzimi di impacchettamento. A diversi livelli di classificazione batterica, VirHost Hunter è più accurato degli strumenti esistenti senza allineamento e rimane affidabile anche quando i virus condividono solo una bassa similarità di sequenza. Valutato su fagi intestinali coltivati con ospiti noti sperimentalmente, identifica gli ospiti corretti con precisione superiore rispetto a un metodo standard basato su CRISPR, e l’uso combinato di entrambi gli approcci migliora ulteriormente i risultati.
Scoprire virus intestinali nascosti legati a malattie
Con il modello calibrato, il team ha applicato VirHost Hunter a un ampio Database di Fagi Intestinali umano che prima aveva informazioni sull’ospite per meno di un terzo delle sue voci. Scansionando proteine della coda e lisine, hanno quasi raddoppiato la quota di fagi con ospiti assegnati e hanno scoperto virus che prendono di mira 29 famiglie di batteri intestinali, molti legati a condizioni croniche come malattie infiammatorie intestinali, malattie cardiache e obesità. In particolare, hanno individuato dozzine di fagi precedentemente non caratterizzati predetti infettare batteri come Akkermansia muciniphila e Prevotella copri, implicati in disturbi autoimmuni e metabolici ma privi di fagi noti.
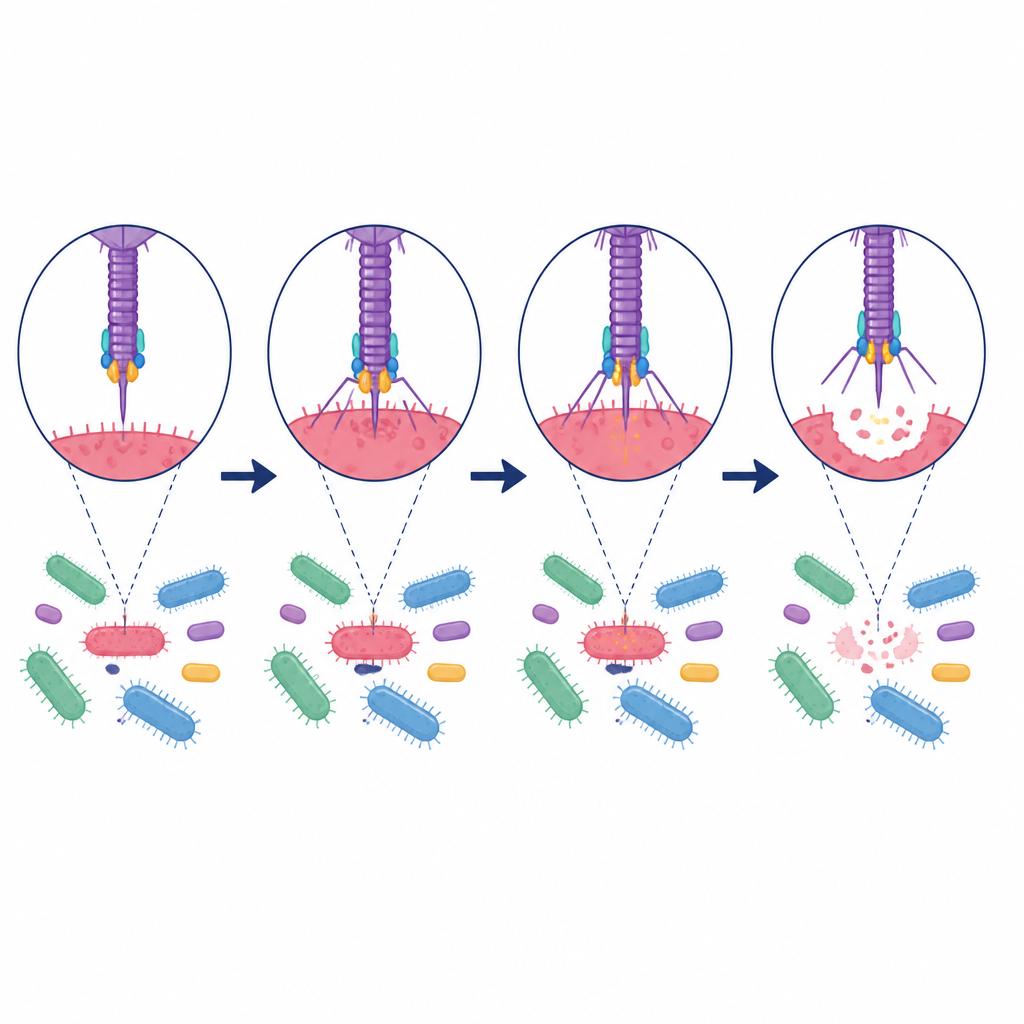
Dalle predizioni digitali a un antimicrobico mirato
Per trasformare queste predizioni in una risorsa pratica, gli autori hanno costruito un Database di Lisine di Fagi Intestinali contenente più di centomila lisine con ospiti intestinali mappati. Hanno esaminato le loro strutture, stabilità e diversità, rivelando molti aggregati distinti e motivi conservati responsabili della rottura della parete batterica. Come prova di principio, hanno selezionato una lisina predetta colpire specificamente Megamonas, un batterio associato all’obesità. Dopo aver sintetizzato questa proteina, hanno mostrato in test di laboratorio che uccide efficientemente Megamonas risparmiando altri comuni microbiani intestinali e ceppi probiotici, dimostrando come l’estrazione guidata dal modello della materia oscura virale possa produrre strumenti altamente selettivi.
Perché questo è importante per la cura futura del microbioma
Questo lavoro dimostra che è possibile collegare un vasto numero di virus intestinali sconosciuti ai loro batteri ospiti usando poche proteine chiave e tecniche moderne di machine learning. Illuminando chi infetta chi nel microbioma, VirHost Hunter potenzia la nostra capacità di esplorare la diversità virale intestinale e di progettare interventi precisi, come lisine su misura, che limitino selettivamente batteri dannosi senza disturbare la comunità microbica più ampia. Pur richiedendo ulteriori test e ingegnerizzazioni prima dell’uso clinico, il quadro fornisce una roadmap potente per trasformare sequenze virali nascoste in strategie mirate per studiare e, un giorno, modulare il nostro ecosistema interno.
Citazione: Du, Z., Li, M., Lin, K. et al. High-resolution phage-host assignment through key proteins using large language models. Nat Commun 17, 4439 (2026). https://doi.org/10.1038/s41467-026-70613-x
Parole chiave: viroma intestinale, batteriofagi, apprendimento automatico, lisine fagiche, terapia del microbioma