Clear Sky Science · pl
Badania śmiertelności syntetycznej związanej z VHL ujawniają CBF-β jako negatywnego regulatora STING
Dlaczego to ma znaczenie dla raka nerki
Rak jasnokomórkowy nerki jest najczęstszą postacią raka nerki, a gdy się rozsiewa, dostępne terapie często nie są w stanie go wyleczyć. Niemal wszystkie te guzy dzielą tę samą wczesną wadę genetyczną: utratę genu nazwanego VHL. Artykuł stawia praktyczne pytanie o dużych implikacjach dla pacjentów: czy można znaleźć drugi słaby punkt, który staje się śmiertelny dla komórki nowotworowej tylko wtedy, gdy brakuje VHL, i jednocześnie uruchomić wewnętrzny alarm przeciwwirusowy organizmu w obrębie guza?
Odnalezienie ukrytej słabości
Naukowcy użyli potężnego podejścia edycji genów — przesiewu CRISPR obejmującego cały genom — w liniach komórek raka nerki, które albo pozbawione były VHL, albo miały go przywróconego. Wyłączając niemal każdy gen po kolei i obserwując, które zmiany zabijały tylko komórki z defektem VHL, poszukiwali «syntetycznych» partnerów VHL: genów, których utrata sama w sobie jest tolerowana, ale w połączeniu z utratą VHL staje się śmiertelna. Spośród wielu kandydatów jeden wyróżniał się w dwóch różnych modelach nowotworowych: gen o nazwie CBFB, kodujący białko znane jako CBF-β, które zwykle współdziała z białkami RUNX w kontrolowaniu aktywności genów. Zespół potwierdził w kilku testach następczych, że usunięcie CBF-β znacząco osłabiało komórki pozbawione VHL w porównaniu z ich odpowiednikami, którym VHL przywrócono. 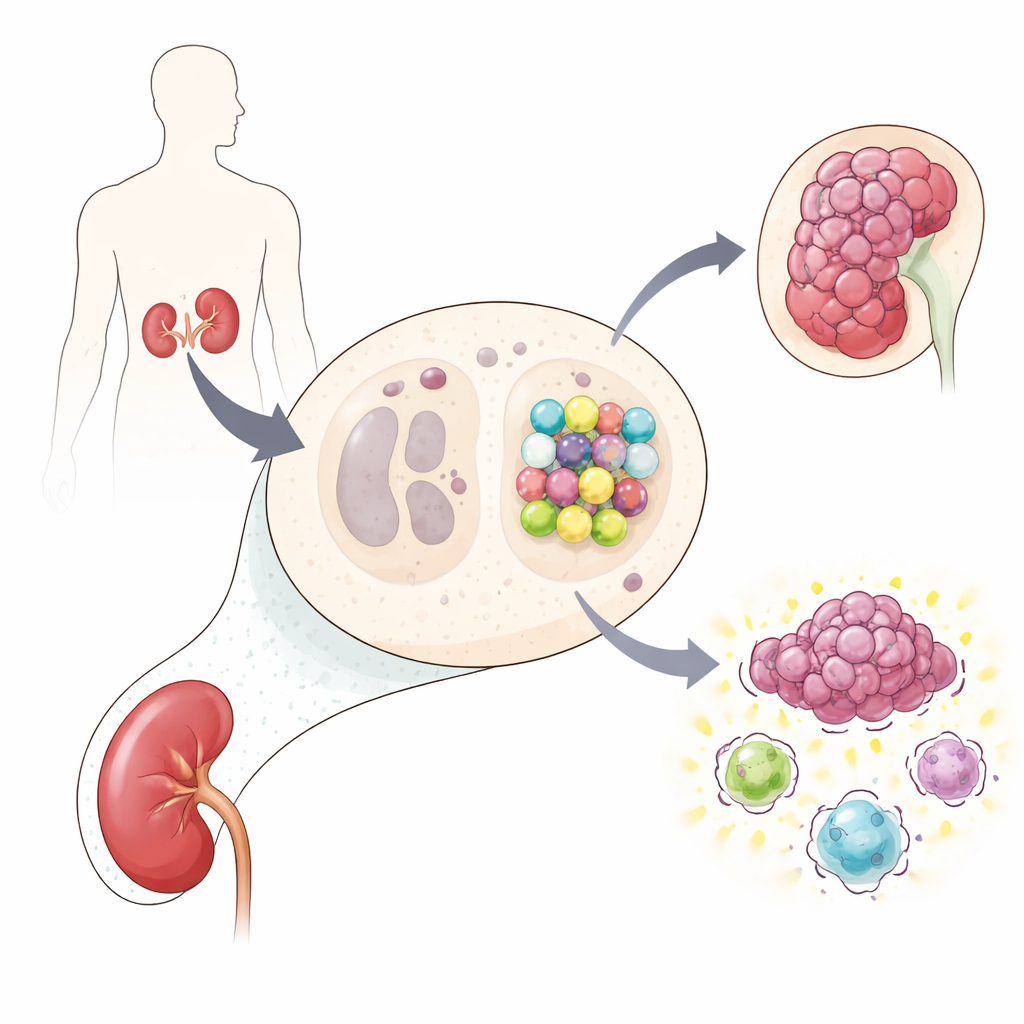
Z płytki Petriego do guzów u myszy
Następnie autorzy sprawdzili, czy ta wrażliwość istnieje w bardziej realistycznych warunkach. Wykazali, że wyczerpanie CBF-β upośledzało wzrost i zdolność do formowania kolonii w kilku liniach raka nerki z defektem VHL, podczas gdy miało znacznie mniejszy wpływ na komórki pochodzące z zdrowej tkanki nerkowej. U myszy ludzkie komórki raka nerki pozbawione CBF-β niemal całkowicie nie tworzyły guzów podskórnych. W modelu ortotopowym, gdzie komórki nowotworowe wszczepiano bezpośrednio do nerek myszy, a następnie indukowano utratę CBF-β, utworzone guzy w dużej mierze przestały rosnąć i rzadko dały przerzuty do płuc. Analizy baz danych pacjentów wykazały, że CBF-β bywa często obfite w ludzkich rakach jasnokomórkowych nerki, a wysokie poziomy jego genu lub białka wiążą się z gorszym przeżyciem pacjentów, co wspiera koncepcję, że komórki nowotworowe są od tego czynnika zależne.
Włączenie wewnętrznego alarmu komórkowego
Aby zrozumieć, dlaczego utrata CBF-β jest tak szkodliwa dla komórek z defektem VHL, badacze porównali profile RNA i białek w komórkach z CBF-β i bez niego. W komórkach pozbawionych VHL usunięcie CBF-β wywołało szeroką odpowiedź przypominającą reakcję przeciwwirusową: wiele genów stymulowanych interferonem, zwykle aktywowanych przy wykrywaniu wirusowego DNA, zostało włączonych. Zaskakująco, ta odpowiedź nie opierała się na klasycznej kaskadzie sygnałowej interferonu przez STAT1 i STAT2. Zamiast tego zależała od bardziej bezpośredniej ścieżki obejmującej adaptor-sensor STING, kinazę TBK1 i czynnik transkrypcyjny IRF3. Po usunięciu CBF-β IRF3 stał się aktywny i pobudził ekspresję genów przeciwwirusowych wewnątrz komórki, nawet bez wykrywalnych fal wydzielanego interferonu.
Zdjęcie hamulca z STING
Badając dalej, zespół ustalił, że CBF-β normalnie działa jako hamulec bezpośrednio na STING. Gdy CBF-β zniknęło, poziomy białka STING i jego mRNA gwałtownie wzrosły, co sprawiło, że komórki stały się znacznie bardziej wrażliwe na fragmenty DNA w cytoplazmie pobudzające szlak cGAS–STING. Transfekcja podwójnego niciowego DNA do komórek deficytowych w CBF-β powodowała dramatyczny wzrost genów przeciwwirusowych i interferonu-β, podczas gdy nadekspresja CBF-β tłumiła tę odpowiedź. Przy użyciu testów wiązania z chromatyną badacze pokazali, że CBF-β wraz z białkami RUNX fizycznie łączy się z genem STING w określonych motywach DNA, bezpośrednio powstrzymując jego aktywność. Nadekspresja RUNX1 zmniejszała sygnaturę STING i mogła przeciwdziałać niektórym następstwom utraty CBF-β, co sugeruje, że to partnerskie działanie transkrypcyjne precyzyjnie reguluje czułość wewnętrznego alarmu immunologicznego. 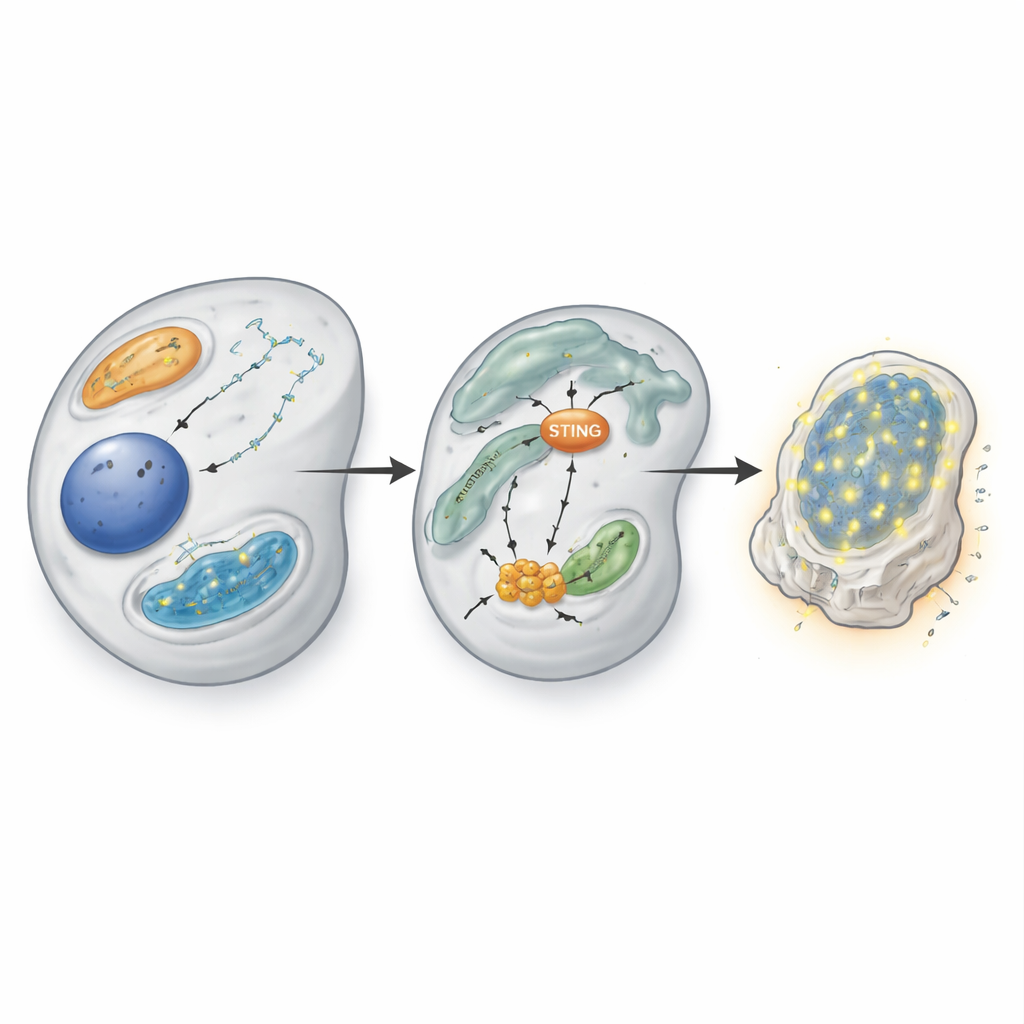
Powiązania z wirusami i nowe terapie
Badanie łączy także ten mechanizm z infekcją wirusową. Białko HIV o nazwie Vif jest znane z wiązania i sekwestracji CBF-β w cytoplazmie, blokując jego normalną współpracę z RUNX w jądrze. Naśladowanie tego przez ekspresję Vif w komórkach raka nerki zwiększało poziomy STING i aktywację genów przeciwwirusowych, podobnie jak usunięcie CBF-β. To wspiera szerszy model, w którym kompleks CBF-β–RUNX działa jako uniwersalny «regulator» odpowiedzi interferonowej napędzanej przez STING, modulując siłę reakcji komórek na nieprawidłowo umieszczone DNA w kontekstach od obrony przeciwwirusowej po raka.
Co to oznacza dla pacjentów
Mówiąc prosto, autorzy identyfikują CBF-β jako dwuzadaniowego pomocnika dla raków nerek z mutacją VHL. Komórki nowotworowe polegają na nim dla przetrwania i wzrostu, ale równocześnie CBF-β przytłumia ich wewnętrzny alarm przeciwwirusowy — skupiony na STING i genach stymulowanych interferonem. Usunięcie CBF-β jednocześnie zabija komórki nowotworowe pozbawione VHL i uwalnia sygnał aktywujący odporność z ich wnętrza. To rodzi możliwość przyszłych terapii przeciwnowotworowych, które celowo zaatakują CBF-β lub jego partnerów RUNX, by wykorzystać mutację VHL, osłabić guz od środka i potencjalnie uczynić go bardziej widocznym oraz podatnym na odpowiedź immunologiczną i immunoterapie.
Cytowanie: Bertlin, J.A.C., Pauzaite, T., Liang, Q. et al. VHL synthetic lethality screens uncover CBF-β as a negative regulator of STING. Nat Commun 17, 3841 (2026). https://doi.org/10.1038/s41467-026-70517-w
Słowa kluczowe: rak jasnokomórkowy nerki, śmiertelność syntetyczna VHL, CBF-beta, szlak STING, interferon typu I