Clear Sky Science · en
VHL synthetic lethality screens uncover CBF-β as a negative regulator of STING
Why this matters for kidney cancer
Clear cell kidney cancer is the most common form of kidney cancer, and once it spreads, current treatments often fail to cure it. Almost all of these tumors share the same early genetic flaw: loss of a gene called VHL. This paper asks a practical question with big implications for patients: can we find another weak spot that becomes fatal to the cancer cell only when VHL is missing, and at the same time flip on the body’s own antiviral alarm system inside the tumor?
Finding a hidden weak spot
The researchers used a powerful gene-editing approach, genome‑wide CRISPR screening, in kidney cancer cell lines that either lacked VHL or had it restored. By turning off almost every gene in the genome, one by one, and watching which changes killed only the VHL‑defective cells, they searched for "synthetic lethal" partners of VHL: genes that are safe to lose on their own but deadly in combination with VHL loss. Among many candidates, one stood out in two different cancer models: a gene called CBFB, which makes a protein known as CBF‑β that normally teams up with RUNX proteins to control gene activity. The team confirmed with several follow‑up tests that removing CBF‑β sharply disadvantaged VHL‑null cells compared with their VHL‑restored counterparts. 
From petri dish to tumors in mice
Next, the authors asked whether this vulnerability exists in more realistic settings. They showed that depleting CBF‑β crippled the growth and colony‑forming ability of several VHL‑defective kidney cancer lines, while having far less impact on cells from healthy kidney tissue. In mice, human kidney cancer cells lacking CBF‑β almost completely failed to form tumors under the skin. In an orthotopic model, where cancer cells were implanted directly into mouse kidneys and then induced to lose CBF‑β, established tumors largely stopped growing and rarely spread to the lungs. Analyses of patient datasets revealed that CBF‑β is often abundant in human clear cell kidney cancers, and high levels of its gene or protein mark patients with poorer survival, supporting the idea that tumor cells depend on this factor.
Turning on the cell’s internal alarm
To understand why CBF‑β loss is so harmful to VHL‑defective cells, the scientists compared RNA and protein profiles in cells with and without CBF‑β. In VHL‑null cells, removing CBF‑β triggered a broad antiviral‑like response: many interferon‑stimulated genes, normally activated when cells detect viral DNA, were switched on. Surprisingly, this response did not rely on the classic interferon signaling relay through STAT1 and STAT2. Instead, it depended on a more direct route involving a sensor adaptor called STING, a kinase called TBK1, and the transcription factor IRF3. When CBF‑β was removed, IRF3 became activated and drove expression of antiviral genes from inside the cell, even without detectable waves of secreted interferon.
Releasing the brake on STING
Diving deeper, the team found that CBF‑β normally acts as a brake on STING itself. When CBF‑β was lost, levels of STING protein and its messenger RNA rose sharply, making cells far more sensitive to bits of DNA in the cytoplasm that feed into the cGAS–STING pathway. Transfecting double‑stranded DNA into CBF‑β‑deficient cells caused a dramatic surge in antiviral genes and interferon‑β, whereas overexpressing CBF‑β dampened this response. Using chromatin‑binding assays, the researchers showed that CBF‑β, together with RUNX proteins, physically associates with the STING gene at specific DNA motifs, directly keeping its activity in check. Overexpressing RUNX1 reduced STING output and could counteract some consequences of CBF‑β loss, suggesting that this transcriptional partnership fine‑tunes the sensitivity of the innate immune alarm. 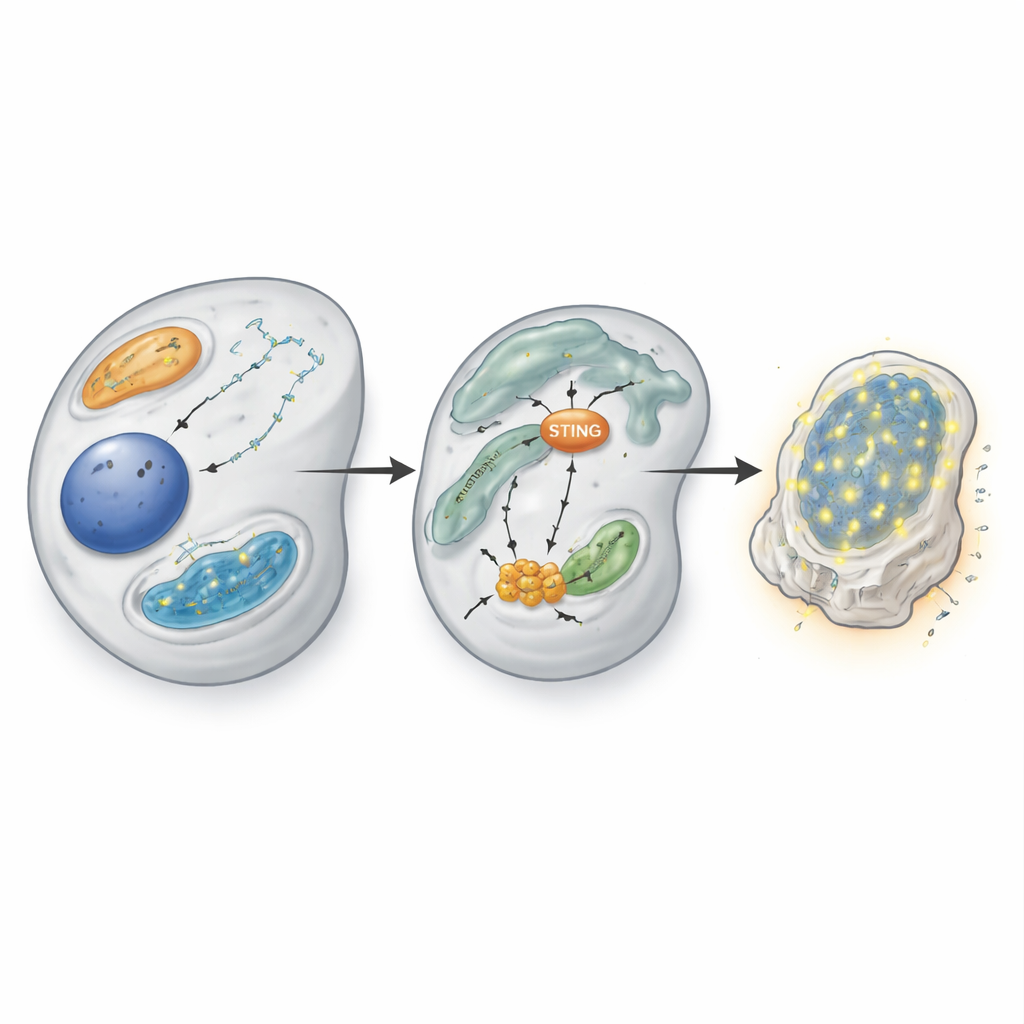
Links to viruses and new therapies
The study also connects this mechanism to viral infection. A protein from HIV, called Vif, is known to bind and sequester CBF‑β in the cytoplasm, blocking its normal work with RUNX in the nucleus. Mimicking this by expressing Vif in kidney cancer cells increased STING levels and antiviral gene activation, similar to deleting CBF‑β. This supports a broader model in which the CBF‑β–RUNX complex serves as a universal "rheostat" on STING‑driven interferon responses, tuning how strongly cells react to misplaced DNA in contexts ranging from viral defense to cancer.
What this means for patients
In simple terms, the authors identify CBF‑β as a double‑edged helper for VHL‑mutant kidney cancers. Tumor cells rely on it for survival and growth, but at the same time CBF‑β keeps their internal antiviral alarm—centered on STING and interferon‑stimulated genes—turned down. Removing CBF‑β both kills VHL‑defective tumor cells and unleashes an immune‑activating signal from within them. This raises the possibility of future cancer therapies that specifically target CBF‑β or its RUNX partners to exploit the VHL mutation, weaken the tumor from the inside, and potentially make it more visible and vulnerable to the immune system and to immunotherapies.
Citation: Bertlin, J.A.C., Pauzaite, T., Liang, Q. et al. VHL synthetic lethality screens uncover CBF-β as a negative regulator of STING. Nat Commun 17, 3841 (2026). https://doi.org/10.1038/s41467-026-70517-w
Keywords: clear cell renal cell carcinoma, VHL synthetic lethality, CBF-beta, STING pathway, type I interferon