Clear Sky Science · pl
Oś FAK/SRC-JNK sprzyja ferroptozie poprzez podwyższanie ekspresji ACSL4
Dlaczego to ma znaczenie dla zdrowia i chorób
Komórki w naszych organizmach mogą ginąć na wiele sposobów, a jednym z nowszych i najbardziej intrygujących jest rodzaj „śmierci napędzanej rdzą” zwany ferroptozą. Proces ten, napędzany przez żelazo i toksyczne lipidy, może być albo silnym sprzymierzeńcem w walce z rakiem, albo bezlitosnym czynnikiem prowadzącym do uszkodzeń narządów. Badanie podsumowane tutaj odkrywa centralny przełącznik regulujący ferroptozę, wyjaśniając, dlaczego niektóre guzy mogą być szczególnie podatne na nowe terapie, a jednocześnie wskazując potencjalny sposób ochrony trzustki w bolesnym i często niebezpiecznym schorzeniu — ostrym zapaleniu trzustki. 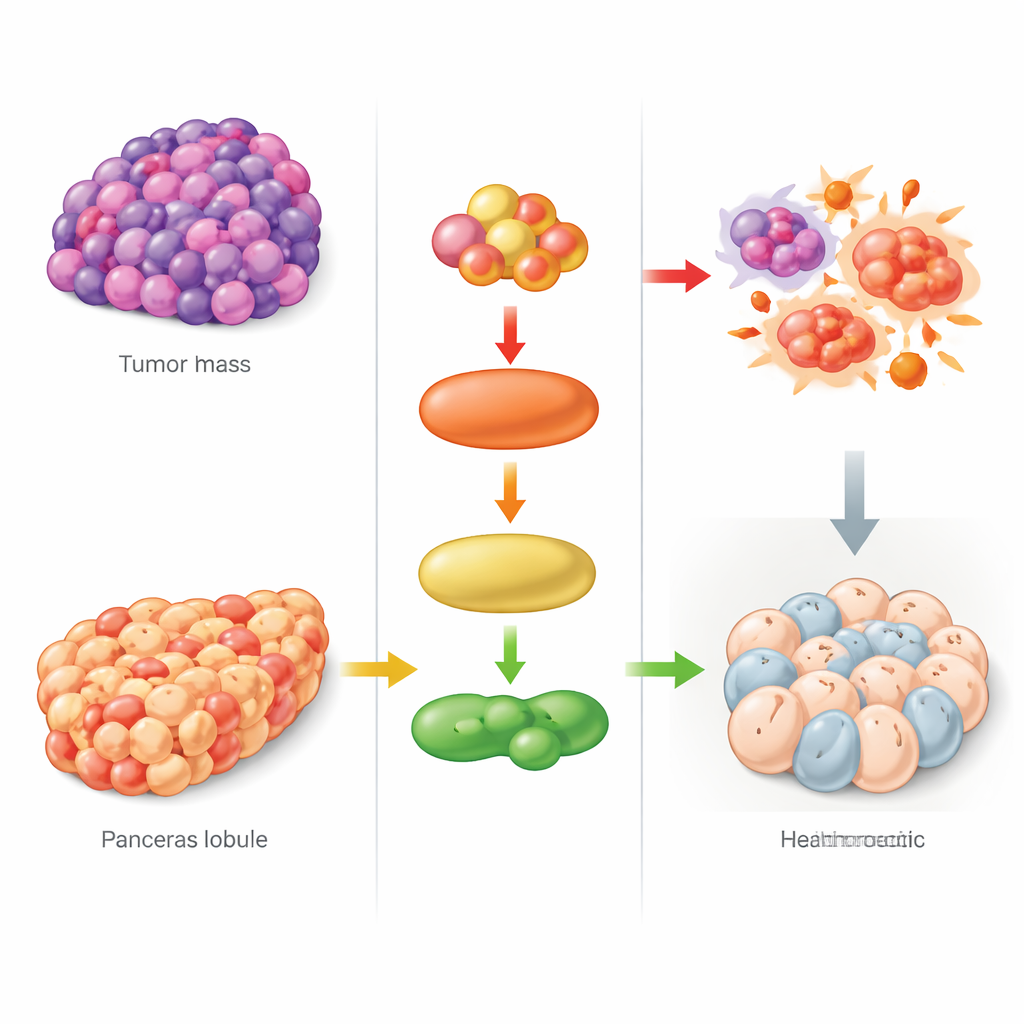
Szczególny rodzaj śmierci komórkowej
Ferroptoza różni się od bardziej znanych form śmierci komórkowej, takich jak apoptoza. Zamiast uporządkowanego rozkładu, komórki ferroptotyczne ulegają zniszczeniu wskutek nagromadzenia uszkodzonych, zoksydowanych lipidów w błonach, napędzanego przez żelazo i reaktywne formy tlenu. Kluczową rolę odgrywa enzym ACSL4, który wprowadza do błon określone, wysoce nietrwałe kwasy tłuszczowe, czyniąc komórki bardziej podatnymi na „zajście”. Gdy mechanizmy komórkowe detoksykujące te lipidy zawodzą, błony ulegają degradacji i komórka zapada się. Ponieważ wiele nowotworów opiera się na zmienionej gospodarce metabolicznej i obróbce żelaza, mogą stać się wyjątkowo wrażliwe na ferroptozę, co czyni ten proces atrakcyjnym celem do eliminacji opornych na leczenie komórek nowotworowych.
Oś sygnałowa, która przygotowuje komórki do śmierci
Naukowcy skupili się na dobrze znanej ścieżce promującej rozwój nowotworów zbudowanej wokół trzech białek: FAK, SRC i JNK. Molekuły te zwykle pomagają komórkom wyczuwać otoczenie, rosnąć i przetrwać, a w zaawansowanych guzach są często nadaktywne. Niespodziewanie, podczas przesiewu tysięcy związków bioaktywnych zespół odkrył, że blokowanie FAK silnie zmniejszało ferroptozę wywoływaną standardowymi induktorami, takimi jak erastyna i RSL3. Szczegółowe eksperymenty w kilku liniach komórkowych nowotworowych wykazały, że zarówno FAK, jak i SRC ulegają aktywacji podczas ferroptozy, a ich wyciszenie — farmakologiczne lub genetyczne — znacząco utrudnia zabicie komórek przez tę ścieżkę. Odwrotnie, wzmocnienie aktywności FAK lub SRC czyni komórki bardziej podatnymi. Brakiem ogniwem okazał się ACSL4: oś FAK/SRC-JNK wyraźnie podnosi poziomy ACSL4, tym samym nasycając błony lipidami sprzyjającymi ferroptozie.
Regulacja w formie popychu i hamowania nad kluczowym enzymem
Pogłębiając badania, autorzy zmapowali, w jaki sposób JNK komunikuje się z DNA komórki, by precyzyjnie regulować produkcję ACSL4. JNK kontroluje sieć czynników transkrypcyjnych — białek wiążących DNA i przełączających geny. Badanie pokazuje, że jedna grupa tych czynników (ATF2, NFATC1, NFATC3 i SMAD4) wiąże się bezpośrednio z genem ACSL4 i zwiększa jego aktywność, promując ferroptozę. Druga grupa (c-Jun, ELK1, HSF1 i STAT3) również wiąże gen ACSL4, lecz tłumi jego ekspresję i hamuje ferroptozę. Testy biochemiczne potwierdzają, że obie grupy czynników zajmują promotor ACSL4, a eksperymenty czasowe sugerują, że „hamulce” uruchamiają się wolniej, stanowiąc opóźnioną informację zwrotną zapobiegającą niekontrolowanemu uszkodzeniu. Ogólnie równowaga między tymi przeciwstawnymi siłami, wszystkie połączone przez oś FAK/SRC-JNK, decyduje o tym, jak łatwo dana komórka ulegnie ferroptozie. 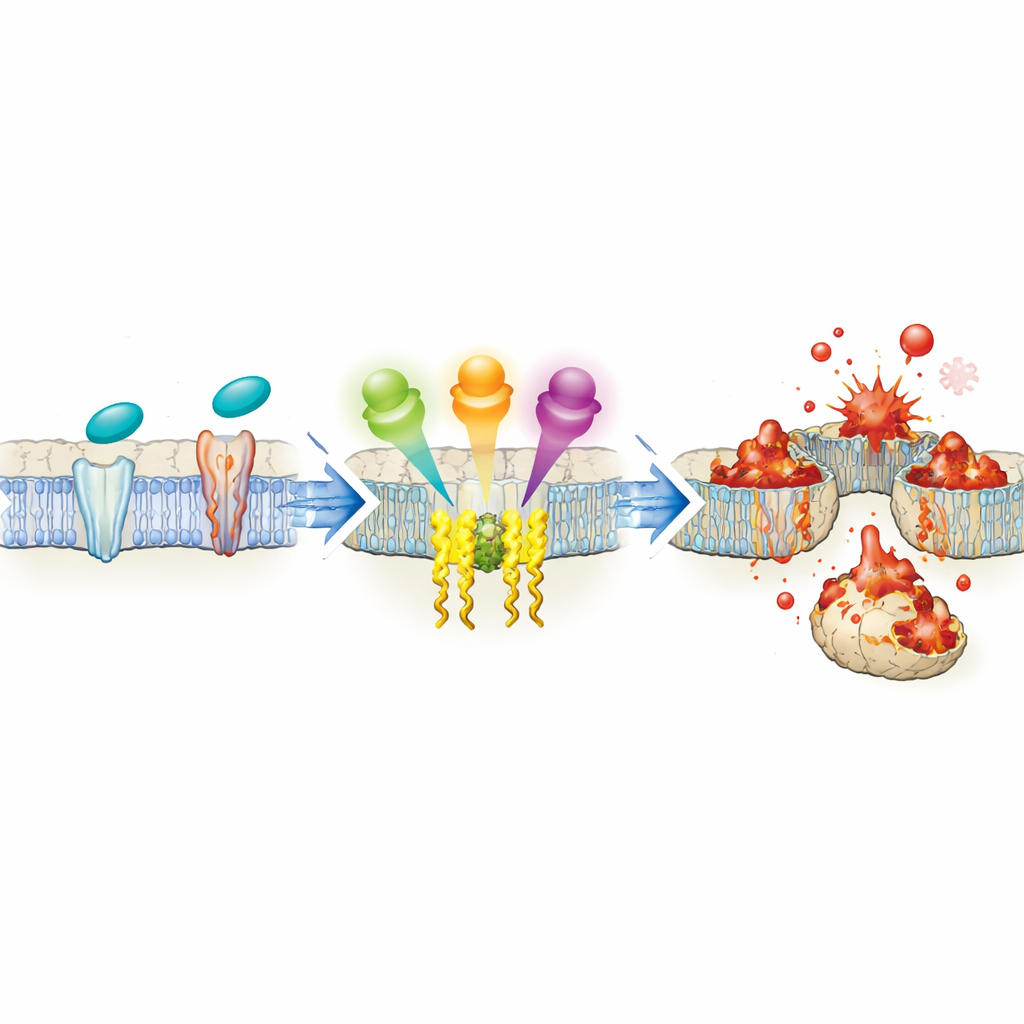
Słabość nowotworów i tarcza trzustki
Podwójny charakter tej ścieżki prowadzi do bardzo odmiennych rezultatów w nowotworach i w zapaleniu. W modelach mysim czerniaka guzy zmodyfikowane tak, by mieć wyższe poziomy FAK, SRC lub JNK, reagowały znacznie lepiej na lek indukujący ferroptozę — kurczyły się bardziej i wykazywały silniejsze biochemiczne oznaki ferroptozy, w tym podwyższony poziom ACSL4 i markerów uszkodzeń lipidów. Guzy z osłabioną aktywnością tej ścieżki opierały się terapii. To sugeruje, że nowotwory uzależnione od sygnalizacji FAK/SRC-JNK mogą mieć wbudowaną „piętę Achillesa”, którą można wykorzystać w terapiach opartych na ferroptozie, a pomiar tej osi sygnałowej mógłby pomóc wytypować pacjentów najprawdopodobniej skorzystających z takiego podejścia. Dla kontrastu, w mysim modelu ostrego zapalenia trzustki wywołanego dużymi dawkami argininy ta sama ścieżka okazała się szkodliwa: była silnie aktywowana, ACSL4 wzrastał, a markery ferroptozy nasilały się w tkance trzustkowej.
Przekucie odkryć w przyszłe terapie
Gdy badacze podawali myszom lub izolowanym komórkom trzustki inhibitory FAK lub SRC, uszkodzenie trzustki, zapalenie i markery związane z ferroptozą wszystkie się zmniejszały, podczas gdy inne formy śmierci komórkowej pozostawały w dużej mierze niezmienione. Wskazuje to, że celowe przyciszenie aktywności osi FAK/SRC-JNK może chronić trzustkę przez ograniczenie ferroptozy, bez szerokiego wyłączania innych programów śmierci. Podsumowując, praca lokuje oś FAK/SRC-JNK-ACSL4 jako główny regulator tej żelazem napędzanej śmierci komórkowej. W guzach jej nadaktywność może zostać wykorzystana, by zwiększyć skuteczność terapii opartych na ferroptozie; w ostrym zapaleniu trzustki i być może w innych chorobach zapalnych, precyzyjnie ukierunkowane inhibitory tej samej ścieżki mogą oferować ochronę. Zrozumienie i modulacja tego molekularnego przełącznika mogą więc otworzyć nowe możliwości zarówno w zabijaniu komórek nowotworowych, jak i w ratowaniu wrażliwych narządów.
Cytowanie: Qin, J., Ma, S., Wang, J. et al. FAK/SRC-JNK axis promotes ferroptosis via upregulating ACSL4 expression. Cell Death Dis 17, 328 (2026). https://doi.org/10.1038/s41419-026-08570-y
Słowa kluczowe: ferroptoza, ACSL4, sygnalizacja FAK SRC JNK, terapia przeciwnowotworowa, ostre zapalenie trzustki