Clear Sky Science · en
FAK/SRC-JNK axis promotes ferroptosis via upregulating ACSL4 expression
Why this matters for health and disease
Cells in our bodies can die in many ways, and one of the newest and most intriguing is a kind of “rust-driven” death called ferroptosis. This process, powered by iron and toxic fats, can be either a powerful ally against cancer or a ruthless driver of organ damage. The study summarized here uncovers a central control switch that tunes ferroptosis up or down, revealing how some tumors may be especially vulnerable to new treatments, while also pointing to a potential way to protect the pancreas in a painful and often dangerous condition: acute pancreatitis. 
A special form of cell death
Ferroptosis is distinct from more familiar forms of cell death like apoptosis. Instead of tidy dismantling, ferroptotic cells succumb to a buildup of damaged, oxidized fats in their membranes, driven by iron and reactive oxygen species. A key player is the enzyme ACSL4, which loads certain highly unstable fats into membranes, making cells easier to “ignite.” When cellular defenses that normally detoxify these fats fail, membranes are destroyed and the cell collapses. Because many cancers rely on altered metabolism and iron handling, they can become unusually sensitive to ferroptosis, making this process an attractive target for killing therapy-resistant tumor cells.
A signaling axis that primes cells to die
The researchers focused on a well-known cancer-promoting pathway built around three proteins: FAK, SRC, and JNK. These molecules usually help cells sense their surroundings, grow, and survive, and they are often overactive in advanced tumors. Unexpectedly, when the team screened thousands of bioactive compounds, they found that blocking FAK strongly reduced ferroptosis triggered by standard inducers such as erastin and RSL3. Detailed experiments in several cancer cell lines showed that both FAK and SRC are switched on during ferroptosis and that turning them down, either with drugs or genetic tools, makes cells much harder to kill by this pathway. Conversely, boosting FAK or SRC activity makes cells more vulnerable. The missing link turned out to be ACSL4: the FAK/SRC-JNK axis sharply raises ACSL4 levels, thereby loading membranes with ferroptosis-prone fats.
Push-and-pull control over a key enzyme
Digging deeper, the authors mapped how JNK communicates with the cell’s DNA to fine-tune ACSL4 production. JNK controls a network of transcription factors—proteins that bind to DNA and switch genes on or off. The study shows that one group of these factors (ATF2, NFATC1, NFATC3, and SMAD4) binds directly to the ACSL4 gene and boosts its activity, thereby promoting ferroptosis. A second group (c-Jun, ELK1, HSF1, and STAT3) also binds the ACSL4 gene but instead dampens its expression and suppresses ferroptosis. Biochemical tests confirm that both sets of factors occupy the ACSL4 promoter region, and timing experiments suggest that the “brakes” engage more slowly, acting as a delayed feedback to avoid runaway damage. Overall, the balance between these opposing forces, all wired through FAK/SRC-JNK, determines how easily a given cell will undergo ferroptosis. 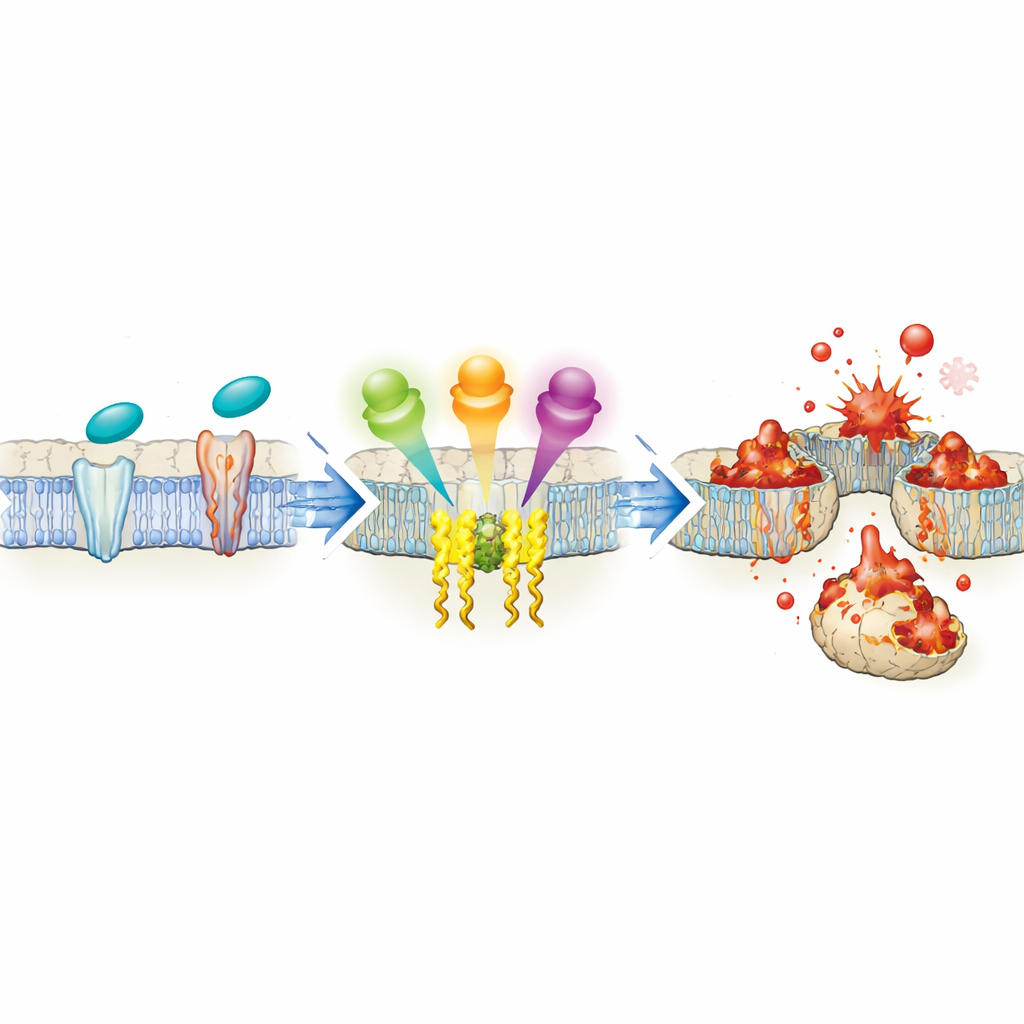
Cancer’s weakness and the pancreas’s shield
The dual nature of this pathway creates very different outcomes in cancer versus inflammation. In mouse melanoma models, tumors engineered to have higher FAK, SRC, or JNK levels responded far better to a ferroptosis-inducing drug, shrinking more and showing stronger biochemical signs of ferroptosis, including elevated ACSL4 and lipid damage markers. Tumors with reduced pathway activity resisted treatment. This suggests that cancers addicted to FAK/SRC-JNK signaling may carry an in-built “Achilles’ heel” that can be exploited by ferroptosis-based therapies, and that measuring this signaling axis could help identify patients most likely to benefit. By contrast, in a mouse model of acute pancreatitis driven by high doses of arginine, the same pathway appeared harmful: it became highly activated, ACSL4 rose, and ferroptosis markers surged in pancreatic tissue.
Turning findings into future therapies
When the researchers treated mice or isolated pancreatic cells with FAK or SRC inhibitors, pancreatic damage, inflammation, and ferroptosis-related markers all decreased, while other forms of cell death remained largely unchanged. This indicates that specifically dialing down FAK/SRC-JNK activity can shield the pancreas by curbing ferroptosis, without broadly shutting off other death programs. Taken together, the work positions the FAK/SRC-JNK-ACSL4 axis as a master regulator of this iron-driven cell death. In tumors, its overactivity may be leveraged to make ferroptosis-based treatments more effective; in acute pancreatitis and possibly other inflammatory diseases, carefully targeted inhibitors of the same pathway might offer protection. Understanding and manipulating this molecular switch could therefore open new avenues for both killing cancer cells and saving vulnerable organs.
Citation: Qin, J., Ma, S., Wang, J. et al. FAK/SRC-JNK axis promotes ferroptosis via upregulating ACSL4 expression. Cell Death Dis 17, 328 (2026). https://doi.org/10.1038/s41419-026-08570-y
Keywords: ferroptosis, ACSL4, FAK SRC JNK signaling, cancer therapy, acute pancreatitis