Clear Sky Science · pl
Oś metabolizmu lipoksygenazy arachidonianowej 5 promująca ferroptozę: potencjalny cel lekooporny w kardiomiopatii wywołanej doksorubicyną
Dlaczego to badanie ma znaczenie dla osób z rakiem
Doksorubicyna to silny i od dawna stosowany lek chemioterapeutyczny, który ratuje życie, ale może też po cichu uszkadzać serce i prowadzić do niewydolności serca wiele lat później. To badanie bada niedawno rozpoznany typ śmierci komórkowej w mięśniu sercowym, zwany ferroptozą, i pyta, czy zablokowanie określonego szlaku kwasów tłuszczowych w komórkach serca mogłoby chronić serce bez osłabiania przeciwnowotworowego działania doksorubicyny.
Lek przeciwnowotworowy, który szkodzi sercu
Doksorubicyna jest powszechnie stosowana w leczeniu nowotworów krwi i guzów litych, jednak jej korzyści ogranicza poważny skutek uboczny: postępujące osłabienie serca, znane jako kardiomiopatia wywołana doksorubicyną. Proponowano wiele mechanizmów uszkodzenia komórek serca, ale ostatnie badania wskazują na ferroptozę — formę śmierci napędzaną przez żelazo i gromadzenie uszkodzonych tłuszczów w błonach komórkowych. W modelach zwierzęcych blokowanie ferroptozy zmniejszało zgony po leczeniu doksorubicyną, co sugeruje, że zrozumienie i zatrzymanie tego procesu mogłoby pomóc pacjentom otrzymywać skuteczną chemioterapię przy mniejszym ryzyku uszkodzenia serca.
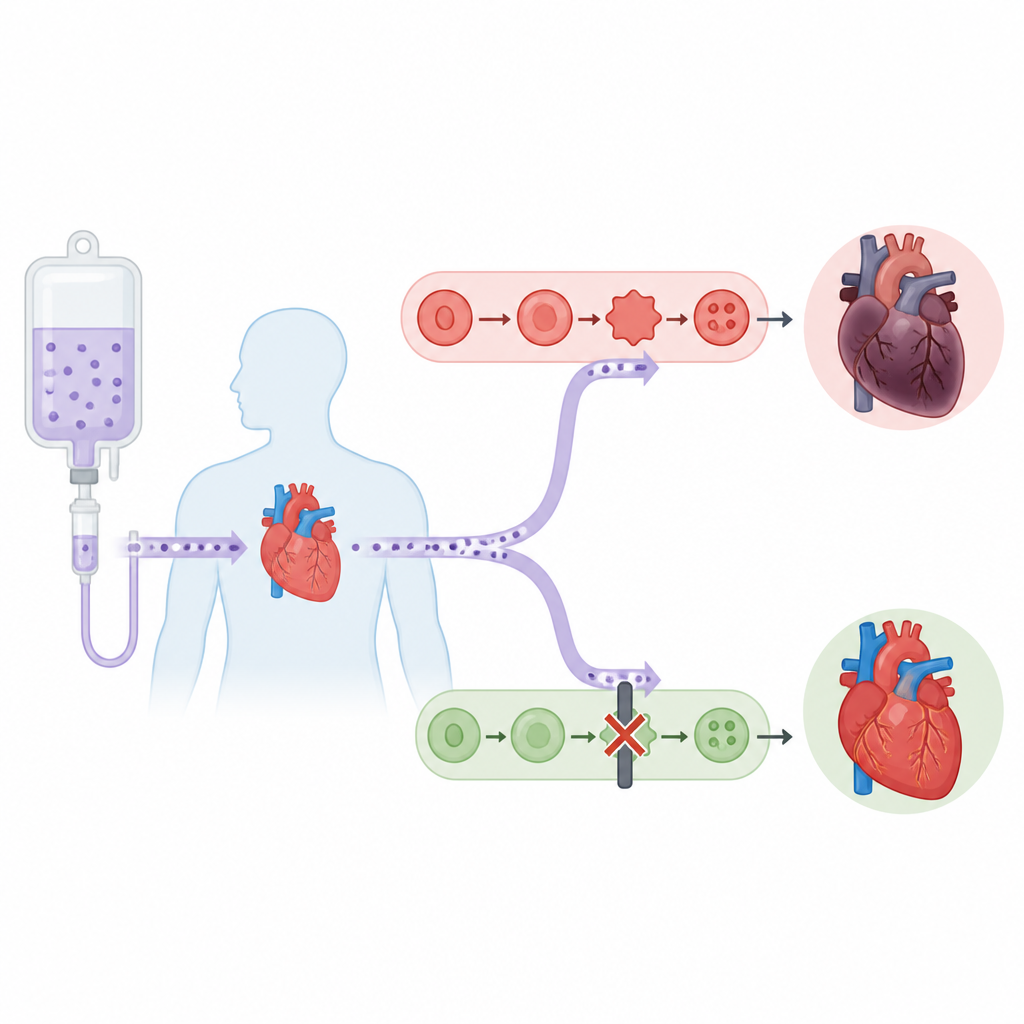
Szlak kwasów tłuszczowych pod podejrzeniem
Autorzy skupili się na enzymie zwanym ALOX5, który normalnie pomaga przekształcać kwas arachidonowy, powszechny składnik budulcowy błon komórkowych, w aktywne lipidowe przekaźniki. Analizując dane dotyczące ekspresji genów oraz badając próbki serca od pacjentów z niewydolnością i od myszy leczonych doksorubicyną, stwierdzili, że ALOX5 i jego produkt 5‑HETE są silnie zwiększone zawsze, gdy serce doświadcza tego rodzaju stresu. W hodowlach komórek przypominających komórki serca i u myszy doksorubicyna zwiększała poziomy ALOX5, łącząc ten szlak tłuszczowy z uszkodzeniami serca związanymi z lekiem.
Włączanie i wyłączanie przełącznika uszkodzeń
Aby sprawdzić związek przyczynowo‑skutkowy, zespół sztucznie zwiększył ekspresję ALOX5 tylko w komórkach mięśnia sercowego myszy. Gdy te myszy otrzymały doksorubicynę, ich serca pompowały mniej wydajnie, wykazywały więcej uszkodzeń strukturalnych i miały wyższe poziomy markerów uszkodzenia we krwi niż zwierzęta kontrolne. Chemiczne wskaźniki ferroptozy, takie jak wyczerpanie glutationu, zwiększona peroksydacja lipidów i obniżone poziomy ochronnych enzymów GPX4 i SLC7A11, również były gorsze. Te szkody łagodził ferrostatyna‑1, bezpośredni inhibitor ferroptozy. W przeciwieństwie do tego, gdy badacze użyli klinicznego inhibitora ALOX5 nazwanego zileutonem lub zastosowali narzędzia genetyczne obniżające ALOX5, struktura serca i jego funkcja skurczowa zostały zachowane, a markery ferroptozy poprawiły się zarówno u myszy, jak i w hodowlach komórek serca.
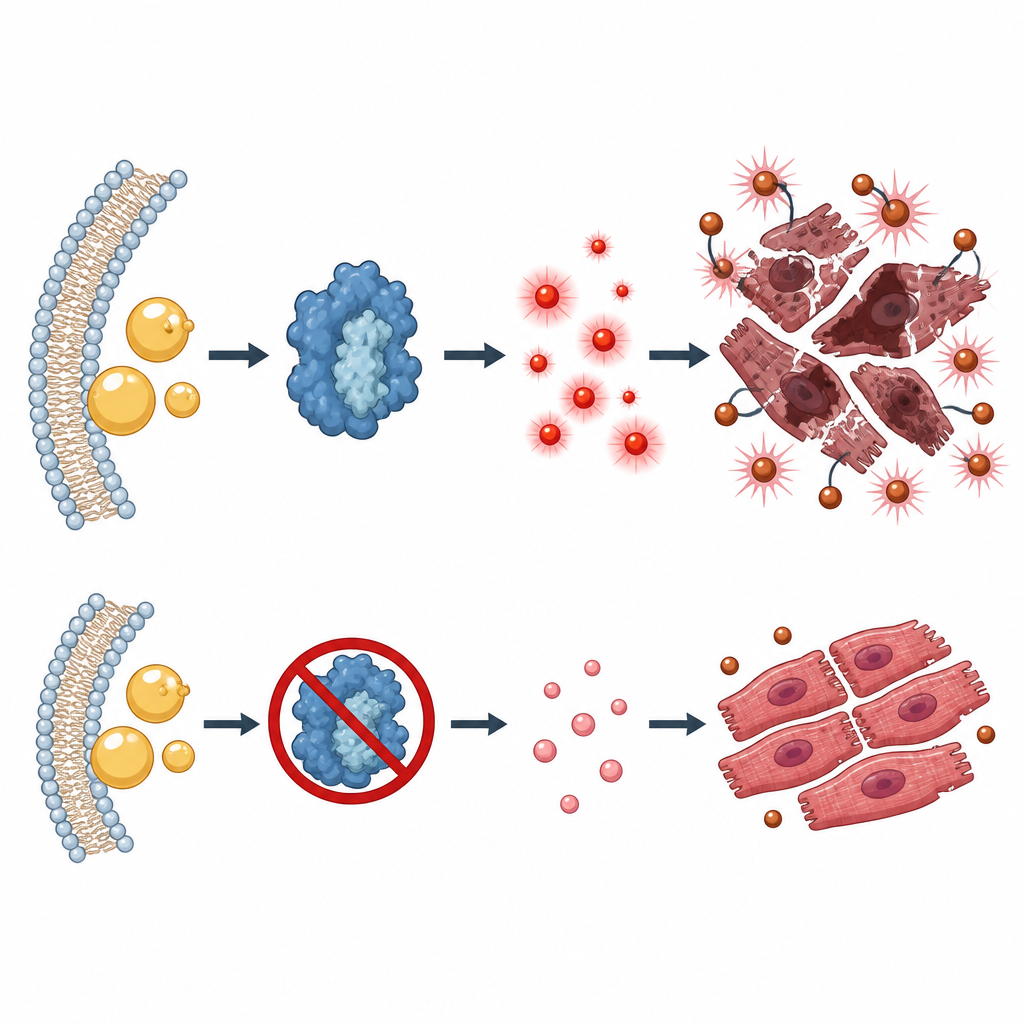
Jak ten szlak popycha komórki serca w stronę ferroptozy
Pogłębiając analizę, badacze wykazali, że suplementacja kwasu arachidonowego, paliwa dla ALOX5, nasilała toksyczne efekty doksorubicyny, podczas gdy blokada ALOX5 odwracała je. Sam produkt lipidowy 5‑HETE był w stanie wywołać zmiany przypominające ferroptozę w komórkach serca, w tym więcej utlenionych tłuszczów i niższe poziomy obrony antyoksydacyjnej komórki. Na poziomie molekularnym 5‑HETE przyspieszał rozkład NRF2 — głównego regulatora, który zwykle włącza wiele genów antyoksydacyjnych — poprzez promowanie jego ubikwitynacji i usuwania przez system proteasomalny komórki. Zjawisko to zależało od szlaku sygnałowego obejmującego PI3K, AKT i GSK‑3β, który działa jak pokrętło regulujące stabilność NRF2.
Ochrona serca bez osłabiania leczenia przeciwnowotworowego
Ponieważ każda strategia ochronna musi zachować przeciwnowotworowe działanie doksorubicyny, zespół sprawdził, czy hamowanie ALOX5 przeszkadza w zabijaniu komórek nowotworowych. W liniach komórkowych raka piersi i wątroby dodanie zileutonu nie osłabiło zdolności doksorubicyny do zmniejszania przeżywalności komórek nowotworowych. Sugeruje to, że blokowanie szlaku ALOX5 może chronić serce przez przywracanie NRF2 i antyoksydacyjnych mechanizmów obronnych komórki, jednocześnie pozwalając doksorubicynie skutecznie atakować komórki nowotworowe.
Co to oznacza dla przyszłej opieki
Mówiąc prościej, badanie identyfikuje „złą chemię” w komórkach serca, w której powszechny kwas tłuszczowy przekształca się w reaktywny produkt, który przyczynia się do spiralnego nasilania się uszkodzeń zależnych od żelaza. Poprzez blokowanie ALOX5 lub jego produktu 5‑HETE, badacze byli w stanie powstrzymać demontaż systemu obronnego serca kierowanego przez NRF2, zmniejszając w ten sposób ferroptozę i uszkodzenie serca w obliczu doksorubicyny. Ponieważ inhibitor ALOX5 jest już zatwierdzony do innych zastosowań, wyniki te stwarzają możliwość, że po starannych badaniach klinicznych ten sam lek mógłby pewnego dnia pomóc pacjentom otrzymywać ratującą życie chemioterapię przy mniejszym ryzyku długoterminowego uszkodzenia serca.
Cytowanie: Chen, L., Sun, X., Zhang, H. et al. Arachidonate lipoxygenase 5 metabolism axis promoting ferroptosis: a potential druggable target for doxorubicin-induced cardiomyopathy. Br J Cancer 134, 1529–1540 (2026). https://doi.org/10.1038/s41416-026-03376-3
Słowa kluczowe: kardiotoksyczność doksorubicyny, ferroptoza, ALOX5, niewydolność serca, zileuton