Clear Sky Science · it
Un test indipendente da firme per le differenze tra gli spettri di mutazione tumorale rivela effetti di carcinogeni e ascendenza
Perché piccole variazioni del DNA contano nel cancro
I tumori crescono perché il loro DNA è stato danneggiato in molti modi piccoli, lasciando dietro di sé schemi distintivi di mutazioni. Questi schemi possono suggerire cosa ha causato il cancro, come il fumo di tabacco, la luce UV o fattori ereditari di rischio. Ma finora i ricercatori non disponevano di un modo semplice e rigoroso per porsi una domanda fondamentale: due gruppi di tumori – per esempio provenienti da persone con diverse ascendenze o da animali esposti a sostanze diverse – hanno davvero schemi di mutazione differenti, oppure le differenze apparenti sono solo rumore? Questo articolo presenta uno strumento statistico di uso generale per rispondere a quella domanda e mostra che può rivelare effetti nascosti di carcinogeni e dell’ascendenza sui genomi tumorali.
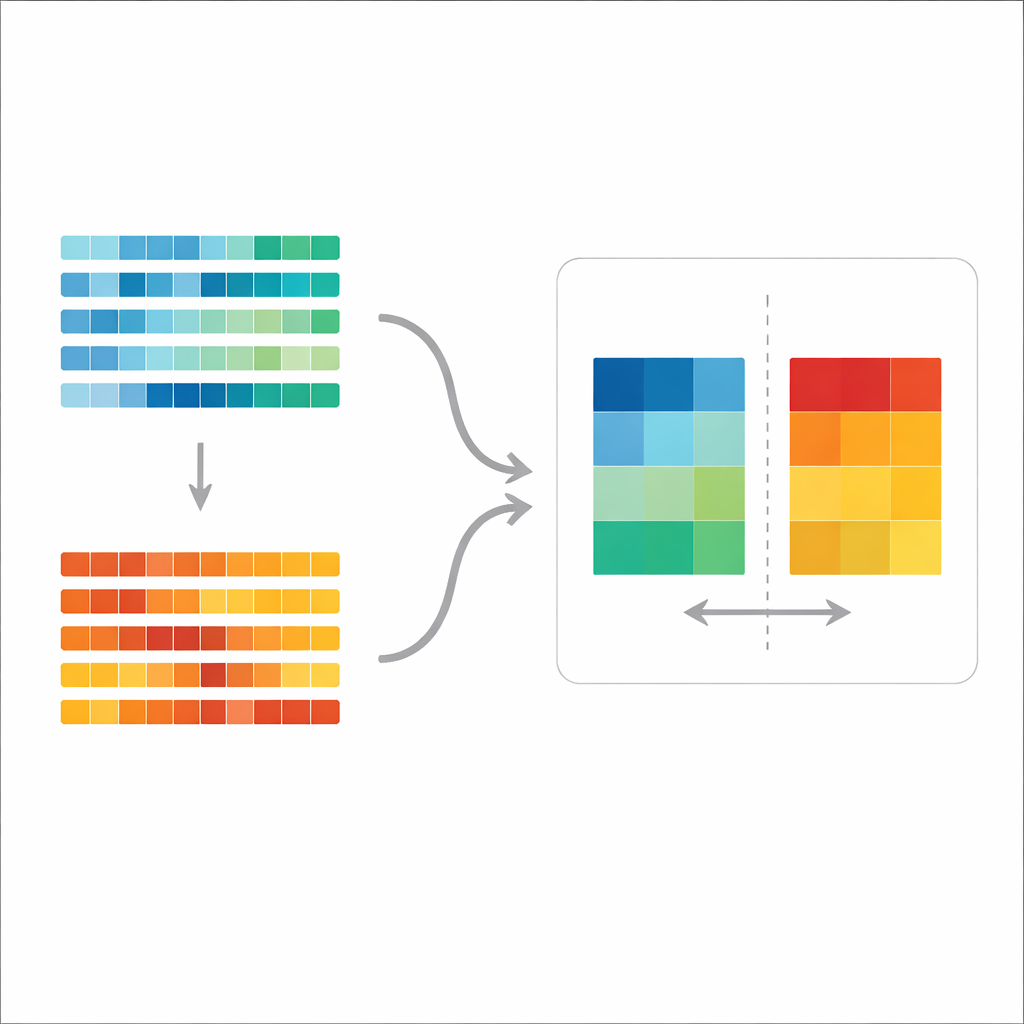
Un nuovo metro per confrontare gli schemi di mutazione
Gli autori partono dall’idea di “spettro mutazionale”, l’insieme complessivo dei tipi di mutazione nel genoma di un tumore. Lavori esistenti spesso scompongono questi spettri in “firme” predefinite considerate rappresentative di processi biologici specifici, quindi esaminano quali firme compaiono in quali tumori. Quel metodo è utile per descrivere i risultati, ma non è ideale per test formali: può essere difficile stabilire se due gruppi differiscono davvero tenendo conto della variazione naturale all’interno di ciascun gruppo e dell’incertezza nell’assegnazione delle firme. Il nuovo metodo, chiamato aggregate mutation spectrum distance (AMSD), evita questo problema lavorando direttamente sugli spettri grezzi e chiedendo, in modo statisticamente controllato, se gli spettri medi di due gruppi sono più diversi di quanto ci si aspetterebbe per caso.
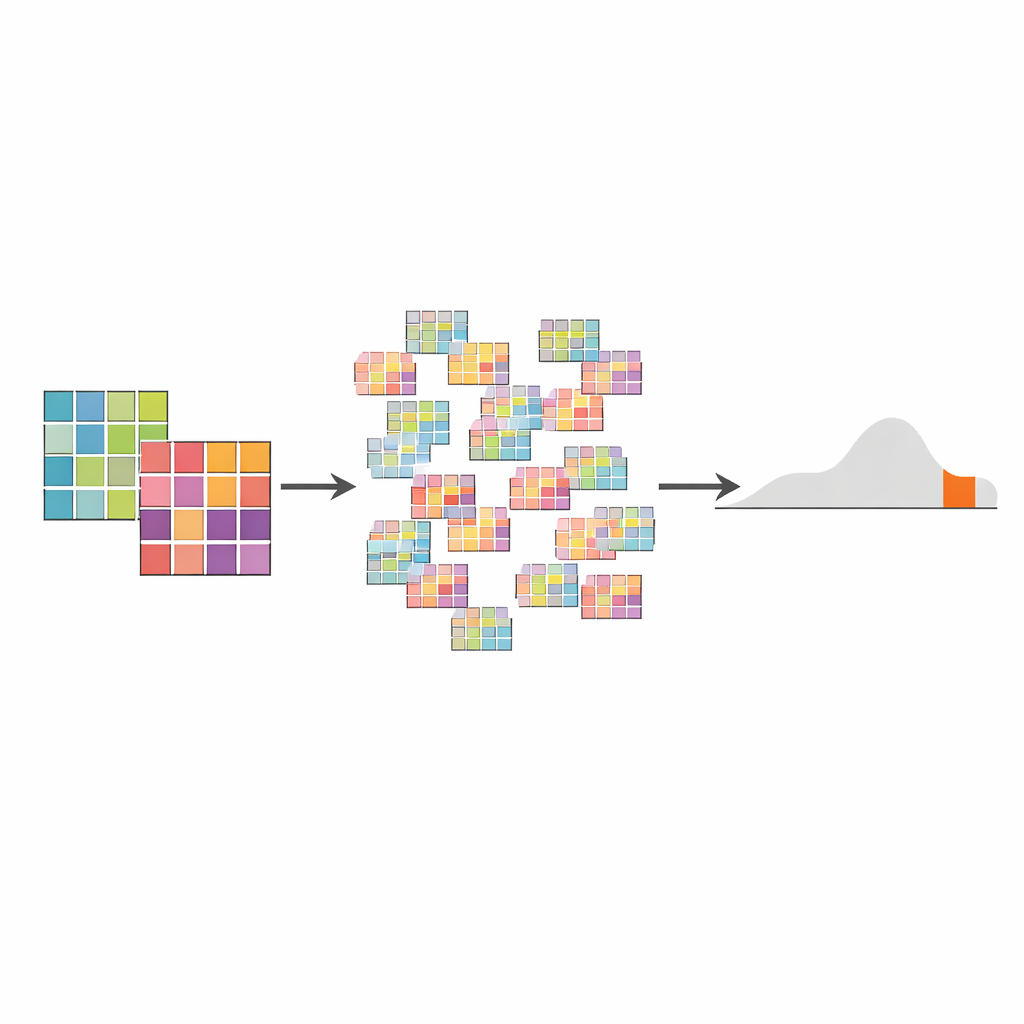
Come funziona il test AMSD sotto il cofano
AMSD combina innanzitutto i dati di mutazione di tutti i tumori in ciascun gruppo in uno “spettro aggregato” per gruppo, dando a ogni tumore lo stesso peso oppure ponderandoli in base al numero di mutazioni che contengono. Misura poi quanto sono distanti questi due spettri usando una misura di distanza come la distanza del coseno, che cattura quanto siano diverse le loro forme. Per decidere se la distanza osservata è significativa, il metodo usa un test di permutazione: rimescola ripetutamente a quale gruppo appartengono i tumori, ricalcola la distanza migliaia di volte e costruisce una distribuzione “null” delle distanze attese se non ci fosse una vera differenza tra i gruppi. Il p-value è semplicemente la frazione di confronto rimescolati che risultano almeno tanto estremi quanto quello reale. Questo approccio gestisce naturalmente dimensioni di campione e conteggi di mutazioni diseguali, e la sua distribuzione nulla può anche suggerire sottotipi nascosti o outlier nei dati.
Effetti nascosti di sostanze cancerogene nei topi
Per mostrare cosa può fare AMSD, il team ha rianalizzato uno studio in cui topi sono stati esposti a 20 diversi sospetti carcinogeni, e i tumori di questi animali sono stati confrontati con tumori di topi di controllo non esposti. Lo studio originale aveva riportato solo tre sostanze chimiche che producevano nuove firme di mutazione inequivocabili. Usando AMSD, gli autori hanno scoperto che 11 dei 20 carcinogeni hanno prodotto spostamenti statisticamente significativi nello spettro complessivo delle mutazioni, anche quando l’analisi classica delle firme non le aveva segnalate. Alcune sostanze, come il 1,2,3-tricloropropano, hanno indotto grandi cambiamenti facilmente riconoscibili. Altre, come il tranquillante oxazepam, hanno prodotto spostamenti più sottili ma coerenti in certi tipi di mutazione, invisibili se si guarda solo alle firme standard. Questi risultati suggeriscono che molti carcinogeni potrebbero non introdurre nuovi tipi di danno, ma piuttosto alterare l’equilibrio dei processi mutazionali già presenti nell’organismo, per esempio modificando la riparazione del DNA o la crescita cellulare.
Schemi di mutazione e ascendenza umana
Gli autori hanno poi analizzato una grande raccolta di tumori umani dal The Cancer Genome Atlas, concentrandosi su tumori in cui c’erano abbastanza casi di persone con ascendenza genetica africana, dell’Asia orientale ed europea. Usando AMSD, hanno confrontato gli spettri di mutazione tra gruppi di ascendenza all’interno di ciascun tipo di tumore. Hanno scoperto differenze significative in 16 dei 67 confronti testati dopo la correzione per test multipli, con sei confronti che rimanevano altamente robusti. Alcuni schemi confermavano risultati precedenti, come una maggiore presenza di un pattern mutazionale legato al fumo nei tumori polmonari di pazienti di ascendenza africana, nonostante questi pazienti riferissero di fumare meno sigarette rispetto a pazienti di ascendenza europea. Altri risultati erano nuovi, inclusa una marcata differenza in una coppia di pattern mutazionali (SBS17a/b) tra pazienti dell’Asia orientale ed europei con carcinoma esofageo, e livelli più alti di certi pattern legati alla polimerasi in pazienti dell’Asia orientale con tumori uterini e del colon-retto. Lo studio sottolinea che queste associazioni con l’ascendenza possono riflettere differenze ambientali, mediche o sociali tanto quanto genetiche, ma rivelano differenze reali nel modo in cui i tumori accumulano mutazioni.
Perché questo strumento cambia il quadro
Nel complesso, queste analisi mostrano che AMSD è un modo sensibile e ampiamente applicabile per rilevare quando e dove gli schemi di mutazione divergono – sia a causa di esposizione chimica, ascendenza o altri fattori. Non sostituisce l’analisi tradizionale delle firme; la completa invece chiedendo prima la domanda pulita “questi gruppi differiscono almeno in qualcosa?” e poi usando le firme per aiutare a spiegare il perché. Lavorando direttamente sugli spettri mutazionali grezzi e minimizzando il numero di test separati, AMSD può scoprire spostamenti sottili ma coerenti che altrimenti verrebbero scartati come rumore. Con l’aumentare e la diversificazione dei dataset sul cancro, questo semplice metro basato sulle permutazioni è pronto ad aiutare i ricercatori a mappare come ambiente, genetica e caso si combinino per modellare le cicatrici del DNA lasciate nei tumori.
Citazione: Hart, S.F.M., Alcala, N., Feder, A.F. et al. A signature-agnostic test for differences between tumor mutation spectra reveals carcinogen and ancestry effects. Commun Biol 9, 462 (2026). https://doi.org/10.1038/s42003-026-09652-5
Parole chiave: spettri mutazionali del cancro, esposizione a carcinogeni, ascendenza genetica, test di permutazione, firme mutazionali