Clear Sky Science · fr
Identification via perturbation d’énergie libre de composés naturels ciblant la thymidylate kinase de Mycobacterium tuberculosis
Pourquoi cette recherche compte pour la tuberculose
La tuberculose reste l’une des maladies infectieuses les plus meurtrières au monde, et la résistance aux médicaments rend son traitement de plus en plus difficile. Cette étude examine comment la nature elle‑même peut offrir de nouveaux plans chimiques pour de futurs médicaments. Plutôt que d’utiliser des animaux de laboratoire ou des patients infectés, les chercheurs ont exploité des simulations informatiques avancées pour parcourir d’immenses bibliothèques de composés végétaux et microbiens afin d’y repérer ceux susceptibles de bloquer une enzyme essentielle à la croissance des bactéries responsables de la tuberculose, ouvrant ainsi de nouvelles pistes pour le développement de médicaments.
Une étape fragile dans la construction de l’ADN bactérien
Les bactéries de la tuberculose dépendent d’une petite enzyme appelée thymidylate kinase pour aider à synthétiser l’ADN nécessaire à leur division et à leur propagation dans l’organisme. Cette enzyme fonctionne comme un ouvrier sur une chaîne de montage, convertissant un fragment constitutif de l’ADN en un autre selon une étape précisément contrôlée. Les cellules humaines possèdent une enzyme apparentée, mais la version bactérienne présente une conformation différente dans des régions cruciales, ce qui permet de la cibler sans nuire à nos propres cellules. Si une petite molécule se coince dans la bonne poche de cette enzyme et bloque son mouvement, la bactérie ne peut plus copier son ADN efficacement, ralentissant ou arrêtant l’infection.
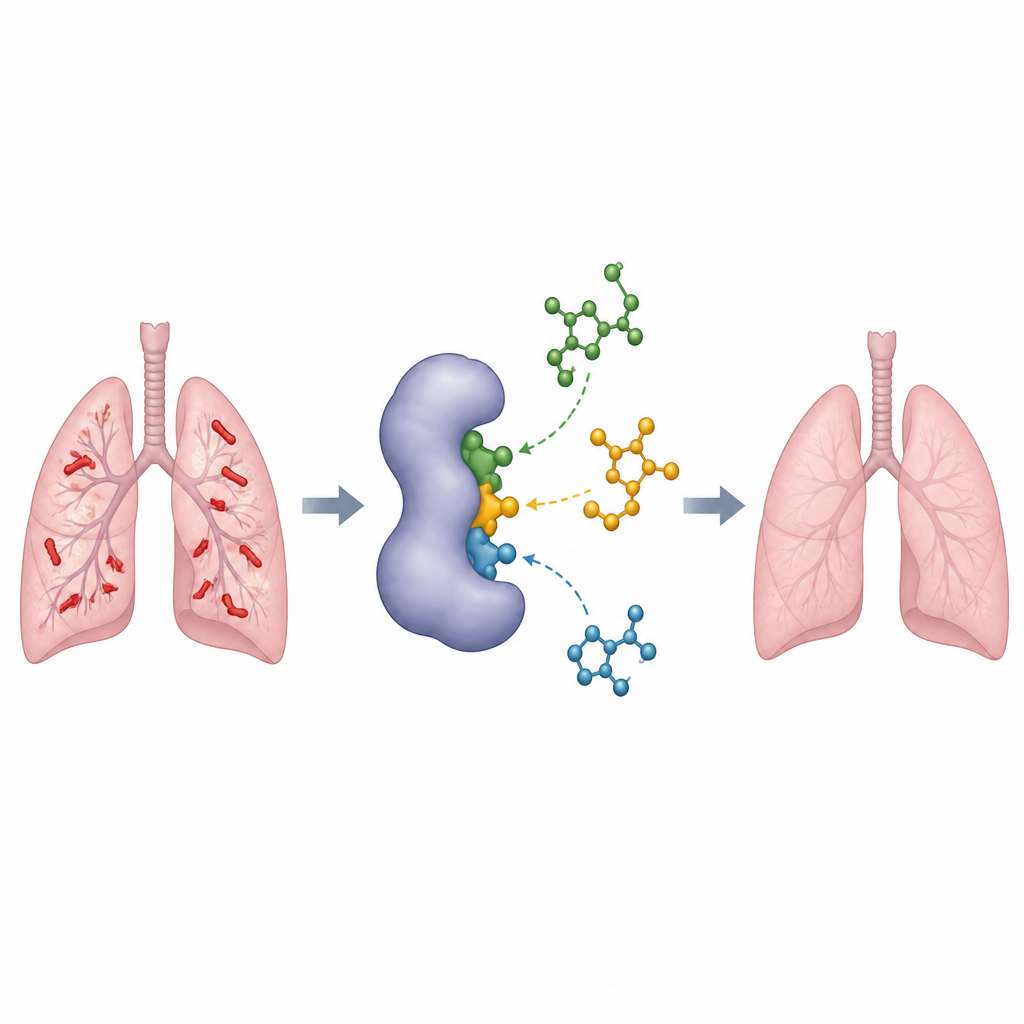
Explorer le catalogue de la nature sur ordinateur
Plutôt que de tester les composés un par un en laboratoire, l’équipe a utilisé des outils informatiques d’aide à la découverte de médicaments pour trier des produits chimiques connus. Ils ont d’abord rassemblé 445 molécules de référence déjà signalées comme interagissant avec cette enzyme et les ont utilisées pour entraîner et valider leurs méthodes. Des programmes de docking sophistiqués ont tenté de « caler » chaque molécule dans la poche active de l’enzyme, estimant la force probable de la liaison. Pour éviter de se tromper, les chercheurs ont aussi docké des milliers de molécules « leurres » ressemblantes qui ne devraient pas se lier, et ont utilisé des tests statistiques pour confirmer que leur protocole de docking distinguait de manière fiable les ligands probables des non‑ligands.
De milliers de candidats à trois molécules remarquables
Avec un pipeline de docking fiable, les scientifiques se sont tournés vers une vaste collection publique de composés naturels appelée base de données COCONUT. Ils ont extrait près de dix mille molécules partageant des caractéristiques structurales avec les meilleurs inhibiteurs de référence, puis les ont affinées à l’aide d’une carte des caractéristiques chimiques clés requises pour la liaison. Des modèles informatiques ont prédit l’absorption, la distribution et l’élimination potentielles de chaque candidat dans l’organisme, et ont signalé ceux susceptibles d’être toxiques. Étape par étape, cet entonnoir a réduit la liste de milliers de molécules à seulement 122 présentant un comportement drug‑like acceptable, puis à trois composés naturels montrant des prédictions de liaison particulièrement fortes et favorables à l’enzyme de la tuberculose.
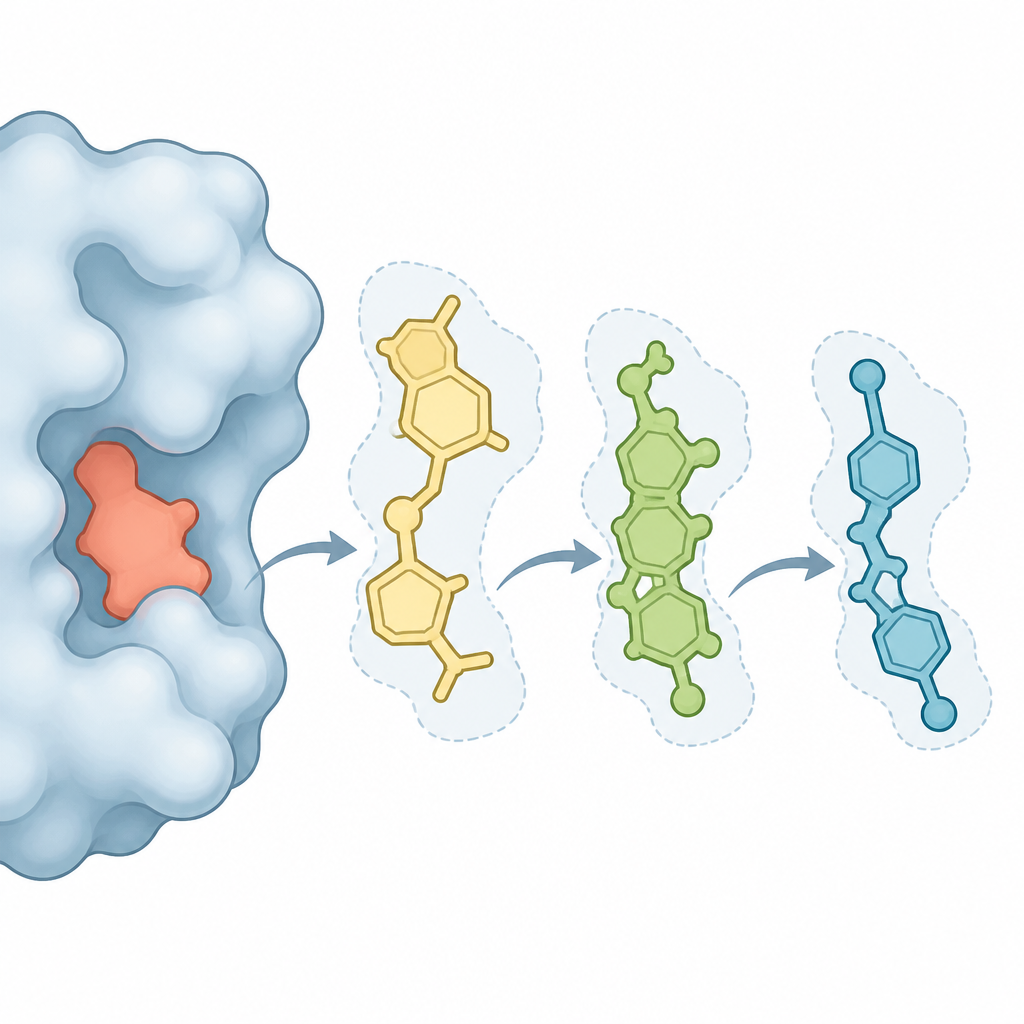
Observer les molécules bouger et se lier en temps virtuel
Pour aller au‑delà des instantanés statiques, l’équipe a exécuté de longues simulations de dynamique moléculaire, qui laissent la protéine et chaque composé candidat évoluer dans une boîte d’eau virtuelle sur une durée de l’ordre de 100 nanosecondes. Ils ont surveillé l’amplitude des fluctuations de la protéine et du ligand, la compacité du complexe et le nombre de contacts stabilisants tels que les liaisons hydrogène formées. Les trois composés naturels sont restés logés dans la même poche enzymatique que celle d’un inhibiteur de référence connu, formant souvent des contacts hydrophobes robustes et des interactions d’empilement avec des acides aminés clés. Des calculs supplémentaires d’énergie libre, visant à estimer l’avantage énergétique de la liaison, ont suggéré qu’un composé en particulier, étiqueté CNP0217487, devrait se lier encore plus fortement que les autres.
Ce que cela signifie pour les futurs traitements contre la TB
Concrètement, les chercheurs ont utilisé des méthodes informatiques puissantes pour repérer des molécules naturelles capables de gripper un engrenage critique de la machine de copie d’ADN de la bactérie tuberculeuse. Parmi des milliers de possibilités, trois composés se sont révélés particulièrement prometteurs, dont un qui semble adhérer à l’enzyme de façon particulièrement forte et stable. Bien que ces résultats soient encore théoriques et doivent être confirmés par des tests biologiques réels, ce travail montre que la diversité chimique de la nature, guidée par des techniques de simulation modernes, peut fournir de nouveaux points de départ pour des médicaments visant la tuberculose résistante aux traitements.
Citation: Chikhale, R.V., Islam, M.A., Suryawanshi, V.S. et al. Free energy perturbation-derived identification of natural compounds targeting Mycobacterium tuberculosis thymidylate kinase. Sci Rep 16, 16272 (2026). https://doi.org/10.1038/s41598-026-46983-z
Mots-clés: tuberculose, produits naturels, inhibiteur enzymatique, criblage virtuel, découverte de médicaments