Clear Sky Science · fr
Structure cristalline par rayons X et étude in silico du diquinoline tétrabromo thio‑ponté incluant des mécanismes anticancéreux via prédiction de cibles et docking moléculaire
Pourquoi une nouvelle molécule anticancéreuse compte
Les médicaments anticancéreux cessent souvent d’être efficaces parce que les cellules tumorales trouvent des moyens de les contourner, notamment lorsque des commutateurs clés de croissance à la surface cellulaire restent actifs. Cet article décrit comment des chimistes et des biologistes se sont associés pour étudier une molécule spécialement conçue qui pourrait aider à inhiber deux de ces commutateurs, connus pour favoriser des cancers agressifs du sein et d’autres localisations. Grâce à des tests informatiques et à l’imagerie cristalline détaillée, ils ont exploré à la fois le potentiel de cette molécule comme médicament et la façon dont ses atomes s’organisent à l’état solide.
Une molécule annulée conçue sur mesure
La recherche porte sur un composé appelé diquinoline tétrabromo thio‑ponté, désigné simplement par composé 3. Il est formé de deux systèmes aromatiques condensés reliés par un atome de soufre et décorés d’atomes de brome, ce qui lui confère une forme rigide, presque en cage. L’équipe a d’abord resynthétisé ce composé en utilisant une réaction organique classique qui assemble de petits blocs en une plus grande boucle. Ils ont confirmé sa composition et sa structure avec des outils analytiques usuels tels que la spectroscopie par résonance magnétique nucléaire et la spectrométrie de masse haute résolution, puis ont utilisé la cristallographie par rayons X pour visualiser l’arrangement des atomes en trois dimensions.
Du empaquetage cristallin au comportement hôte–invité
Lors de la cristallisation depuis le p‑xylène, le composé 3 a formé un complexe hôte–invité dans lequel deux molécules hôtes entourent une seule molécule invitée de solvant. L’analyse par rayons X a montré que les hôtes s’assemblent en dimères via un réseau d’attractions faibles : interactions entre les atomes de brome et l’azote, l’hydrogène et le soufre, ainsi que des liaisons hydrogène subtiles. Ces forces non covalentes créent un motif répétitif qui emprisonne le p‑xylène à l’intérieur. Cette « étreinte » supramoléculaire illustre comment la forme du composé et la présence d’hétéroatome favorisent la reconnaissance et la rétention d’autres molécules, un comportement pertinent pour le stockage, la délivrance ou l’organisation des médicaments dans l’organisme.
Sonde du comportement médicamenteux par ordinateur
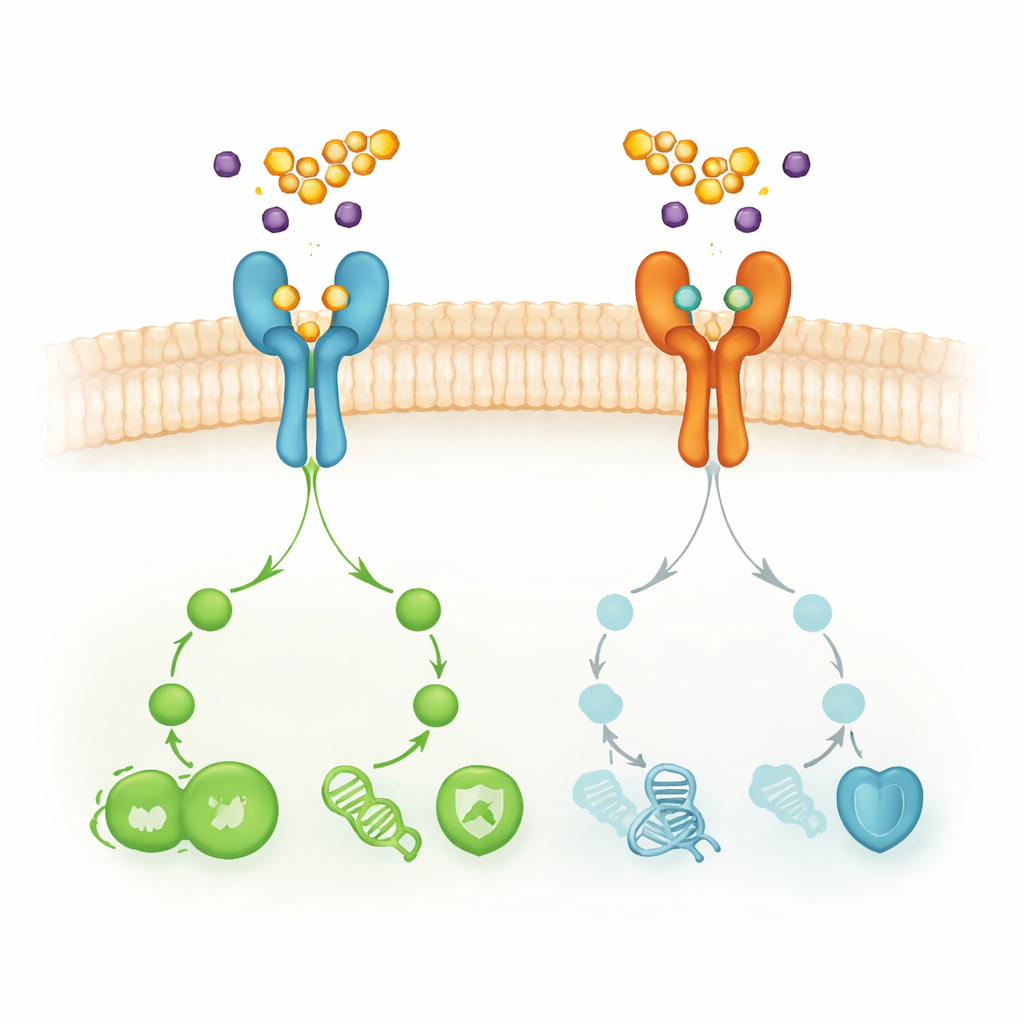
Pour juger si le composé 3 pourrait fonctionner comme médicament, les auteurs ont utilisé une batterie d’outils in silico, c’est‑à‑dire informatiques. Ils ont prédit quelles protéines humaines le composé est le plus susceptible de cibler et comment il pourrait être absorbé, distribué, métabolisé et éliminé. La prédiction de cibles a mis en évidence 36 protéines possibles, avec deux récepteurs de croissance à la surface cellulaire — EGFR et HER2 (également appelé ERBB2) — qui émergent comme des nœuds centraux dans un réseau d’interactions dense. Ces récepteurs sont bien connus pour leurs rôles dans les cancers du sein, du poumon et d’autres, et pour contribuer à la résistance lorsque les médicaments actuels bloquant EGFR deviennent inefficaces.
Relier le composé aux voies de signalisation du cancer
L’analyse des voies de signalisation a placé les protéines cibles prédites du composé 3 dans plusieurs circuits liés au cancer, y compris les voies générales du cancer, la signalisation PI3K–Akt, l’angiogenèse pilotée par le VEGF, et, notablement, la résistance aux inhibiteurs d’EGFR et la signalisation ERBB. Des simulations de docking moléculaire ont ensuite modélisé l’ajustement du composé 3 dans les sites actifs d’EGFR et de HER2. Les résultats suggèrent une liaison stable aux deux récepteurs, avec une affinité prédite plus élevée pour HER2. Dans ces modèles, le composé forme plusieurs liaisons hydrogène et contacts hydrophobes avec des résidus d’acides aminés clés, ce qui laisse penser qu’il pourrait perturber physiquement la capacité des récepteurs à transmettre les signaux de croissance à l’intérieur de la cellule.
Des promesses tempérées par des inquiétudes de sécurité
Les mêmes outils informatiques ont aussi soulevé des signaux d’alerte. Bien que le composé 3 respecte des règles usuelles de « drug‑likeness » et semble peu susceptible d’affecter le cerveau ou les canaux responsables du rythme cardiaque, il est prédit comme étant mal absorbé par l’intestin et fortement lié aux protéines sanguines, ce qui limite la fraction libre circulante du médicament. Plus sérieusement, plusieurs modèles indiquent un risque de lésions hépatiques et de dommages à l’ADN, ainsi qu’une forte interférence avec des enzymes responsables du métabolisme de nombreux autres médicaments. Ces éléments suggèrent que, sans optimisation chimique supplémentaire, le composé pourrait entraîner de la toxicité ou des interactions médicamenteuses problématiques.
Ce que nous apprend ce travail
Globalement, l’étude présente le composé 3 comme un point de départ intrigant plutôt que comme un médicament prêt à l’emploi. Sa forme et ses caractéristiques électroniques lui permettent de former des cristaux hôte–invité ordonnés et de se lier in silico à des récepteurs moteurs du cancer comme EGFR et HER2, soutenant l’idée qu’il pourrait aider à combattre des tumeurs résistantes aux traitements. Dans le même temps, les problèmes prédits d’absorption et de sécurité signifient que les chimistes devront repenser et affiner la structure, suivis d’essais expérimentaux et animaux rigoureux. Pour l’heure, ce travail fournit une cartographie détaillée de la manière dont une molécule soigneusement conçue pourrait un jour rejoindre la lutte contre des cancers difficiles à traiter — et rappelle que même des candidats prometteurs doivent franchir de nombreux obstacles avant de devenir des thérapies cliniques.
Citation: Alshahateet, S.F., Al-Mazaideh, G.M., Al-Trawneh, S.A. et al. X-ray crystal structure and in silico investigation of Tetrabromo thia-bridged diquinoline including anticancer mechanisms via target prediction and molecular docking. Sci Rep 16, 13094 (2026). https://doi.org/10.1038/s41598-026-42845-w
Mots-clés: inhibiteurs EGFR HER2, docking moléculaire, résistance aux médicaments en cancer, hôte‑invité supramoléculaire, ADMET profiling