Clear Sky Science · fr
Modèle cinétique multi-échelle de l’oligomérisation de l’éthylène dans le métal-organique Ni-NU-1000
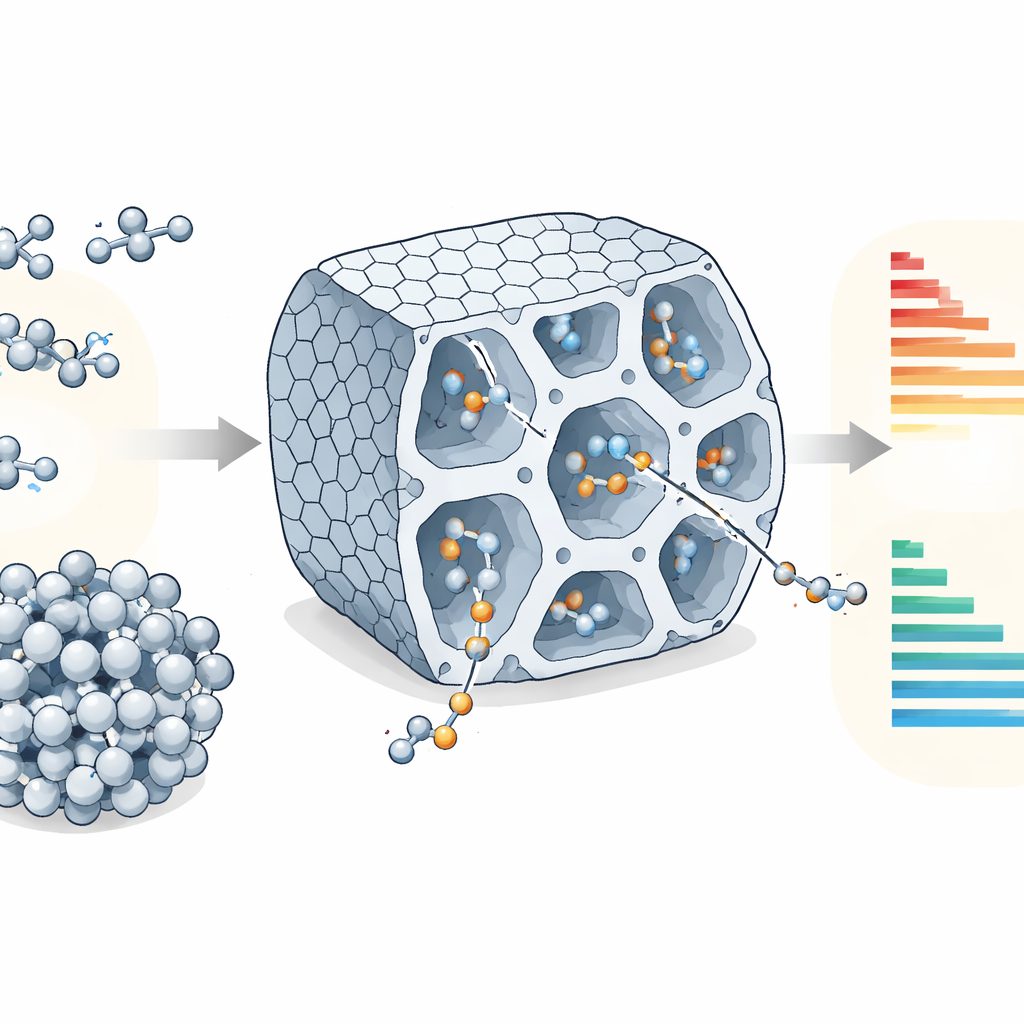
Transformer un gaz simple en briques sur mesure
L’éthylène, l’une des molécules les plus élémentaires produites en grande quantité par l’industrie chimique, peut être assemblé en chaînes plus longues qui servent d’ingrédients pour des plastiques, des détergents et de nombreux produits du quotidien. Mais l’industrie ne veut pas seulement « plus » de produit ; elle veut des chaînes de longueurs très spécifiques. Cet article montre comment la modélisation informatique peut prédire et ajuster quelles longueurs de chaîne se forment à l’intérieur d’un catalyseur poreux solide, guidant potentiellement la conception de procédés chimiques plus propres et plus efficaces.
Pourquoi la forme d’un catalyseur compte
Les chimistes se concentrent souvent sur les atomes métalliques qui pilotent les réactions, mais dans les matériaux poreux, l’échafaudage environnant peut être tout aussi déterminant. Ici, le métal est le nickel, ancré sous forme d’atomes isolés dans un cadre métal–organique nommé NU-1000. Ce réseau ressemble à une éponge faite de tunnels ordonnés et de petites pièces : des canaux larges permettent aux molécules de se déplacer, tandis que des cavités plus petites hébergent les sites de nickel qui relient les molécules d’éthylène en courtes chaînes appelées oligomères. Des études antérieures ont montré que ce matériau peut produire des composés de valeur tels que des butènes et des hexènes, mais il restait flou de savoir comment l’interaction entre la chimie intrinsèque de la réaction et la structure des pores contrôle les produits dominants.
Relier les événements atomiques au comportement du réacteur
Les auteurs construisent un modèle cinétique multi-échelle qui relie les processus de l’échelle atomique à celle du réacteur. D’abord, des calculs de mécanique quantique fournissent les barrières énergétiques pour chaque étape élémentaire de la réaction sur le site de nickel : l’éthylène s’adsorbe, s’insère dans une chaîne en croissance, puis finit par se détacher en tant que molécule achevée. Ensuite, de vastes simulations moléculaires décrivent comment l’éthylène et ses produits s’adsorbent à l’intérieur des pores et à quelle vitesse ils diffusent à travers le réseau. Ces éléments alimentent un modèle d’équation maître qui suit les concentrations dépendantes du temps de toutes les espèces aux températures et pressions réalistes, pour des réacteurs en flux continu comme pour des réacteurs batch fermés.
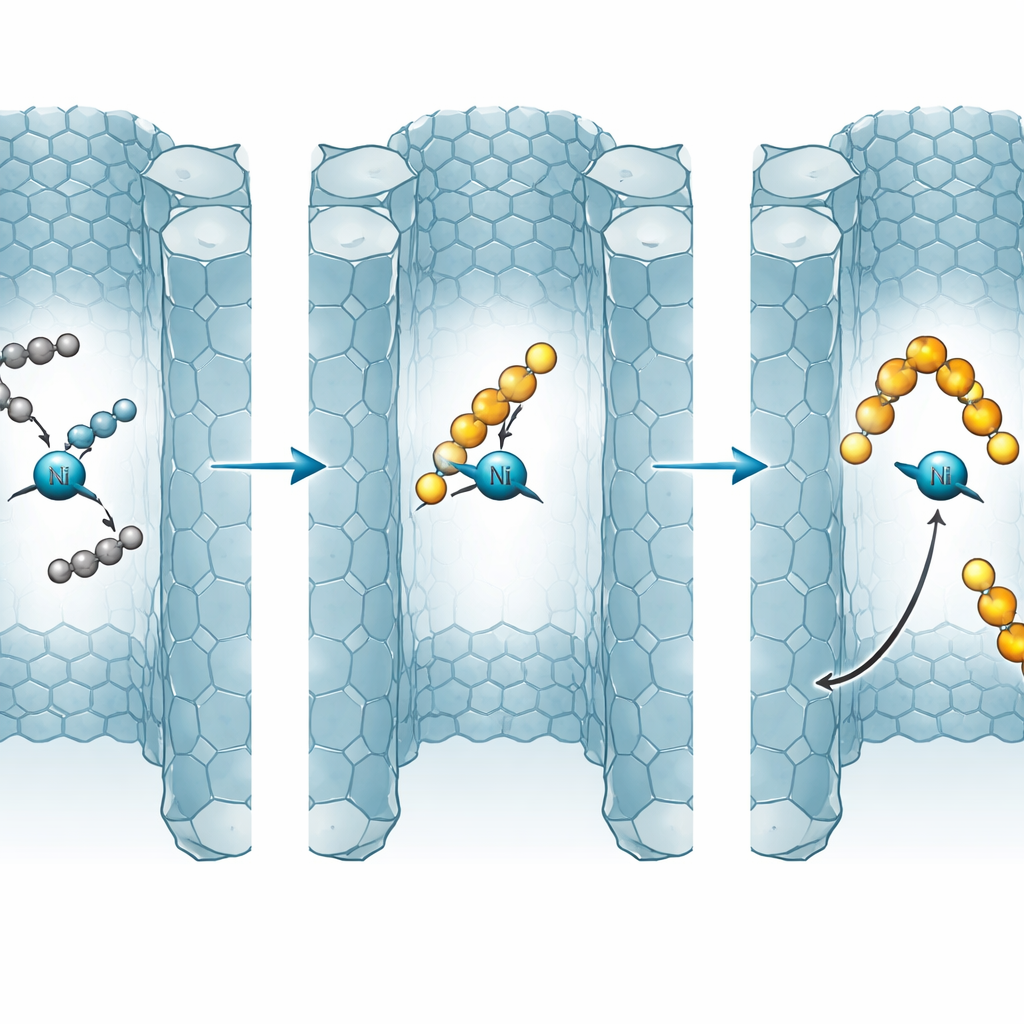
Comment les chaînes croissent, s’arrêtent et se déplacent
À l’intérieur du NU-1000, l’éthylène s’ajoute à une liaison nickel–carbone par étapes répétées, allongeant la chaîne. À tout moment, une voie concurrente peut « terminer » la croissance, libérant un oléfine et régénérant le site de nickel. Le modèle montre que l’équilibre entre croissance et terminaison est très sensible à la température, à la pression et à la facilité avec laquelle les molécules peuvent diffuser hors des pores. À des températures modérées, le système favorise la formation de chaînes à quatre carbones, offrant une fenêtre de haute sélectivité pour les butènes. Lorsque la température augmente davantage, la terminaison et les réactions inverses s’accélèrent, et les chaînes plus longues deviennent plus stables avec le temps, poussant la distribution vers des produits plus lourds qui peuvent finir par ressembler à des cires ou à des polymères.
Quand la diffusion devient un levier caché
Un aperçu clé est que le « temps de séjour » dans les pores agit comme un bouton de contrôle supplémentaire. Dans de petites particules aux courtes distances de diffusion, les produits nouvellement formés s’échappent rapidement, les gelant efficacement à de courtes longueurs de chaîne et maintenant le catalyseur disponible. Dans des particules plus grandes ou des lits peu compactés, les produits persistent, sont plus susceptibles de se ré-adsorber et peuvent croître en chaînes plus longues avant de partir. Le modèle prédit que l’augmentation de la longueur de diffusion effective ou l’augmentation de la charge en nickel resserre ou même efface la fenêtre de sélectivité pour les butènes, conduisant à des oligomères plus lourds et à un risque accru de colmatage des pores et de désactivation du catalyseur, en particulier en exploitation en flux. L’exploitation en batch, où les produits ne sont pas entraînés, favorise naturellement encore plus fortement les produits lourds.
Règles de conception pour des catalyseurs poreux plus intelligents
En unissant structure électronique, adsorption, diffusion et conditions de réacteur dans un seul cadre, ce travail explique pourquoi des sites de nickel similaires peuvent se comporter très différemment selon l’hôte poreux et le mode d’exploitation. Pour le NU-1000 à base de nickel, la recette la plus prometteuse pour fabriquer sélectivement des oléfines à chaîne courte combine de petites tailles de particules effectives, une charge en nickel modérée et des configurations de réacteur qui éliminent rapidement les produits. Plus généralement, l’étude démontre que contrôler la façon dont les molécules se déplacent à travers et se disputent l’espace dans les catalyseurs poreux est tout aussi crucial que le réglage du site métallique actif lui-même, offrant une stratégie transposable pour concevoir des matériaux de nouvelle génération qui transforment des matières premières simples en produits précisément ciblés.
Citation: Avdoshin, A., Matsokin, N.A., Huynh, TN. et al. Multiscale kinetic model of ethylene oligomerization in Ni-NU-1000 metal-organic framework. npj Comput Mater 12, 124 (2026). https://doi.org/10.1038/s41524-026-02044-7
Mots-clés: oligomérisation de l’éthylène, matériaux métal-organiques, catalyse à atome isolé, diffusion et transport de masse, modélisation de la sélectivité catalytique