Clear Sky Science · fr
Le séquençage séquentiel révèle l’architecture et la complexité des variants génomiques chez des patients atteints du syndrome d’Alport
Pourquoi cette étude sur les reins est importante
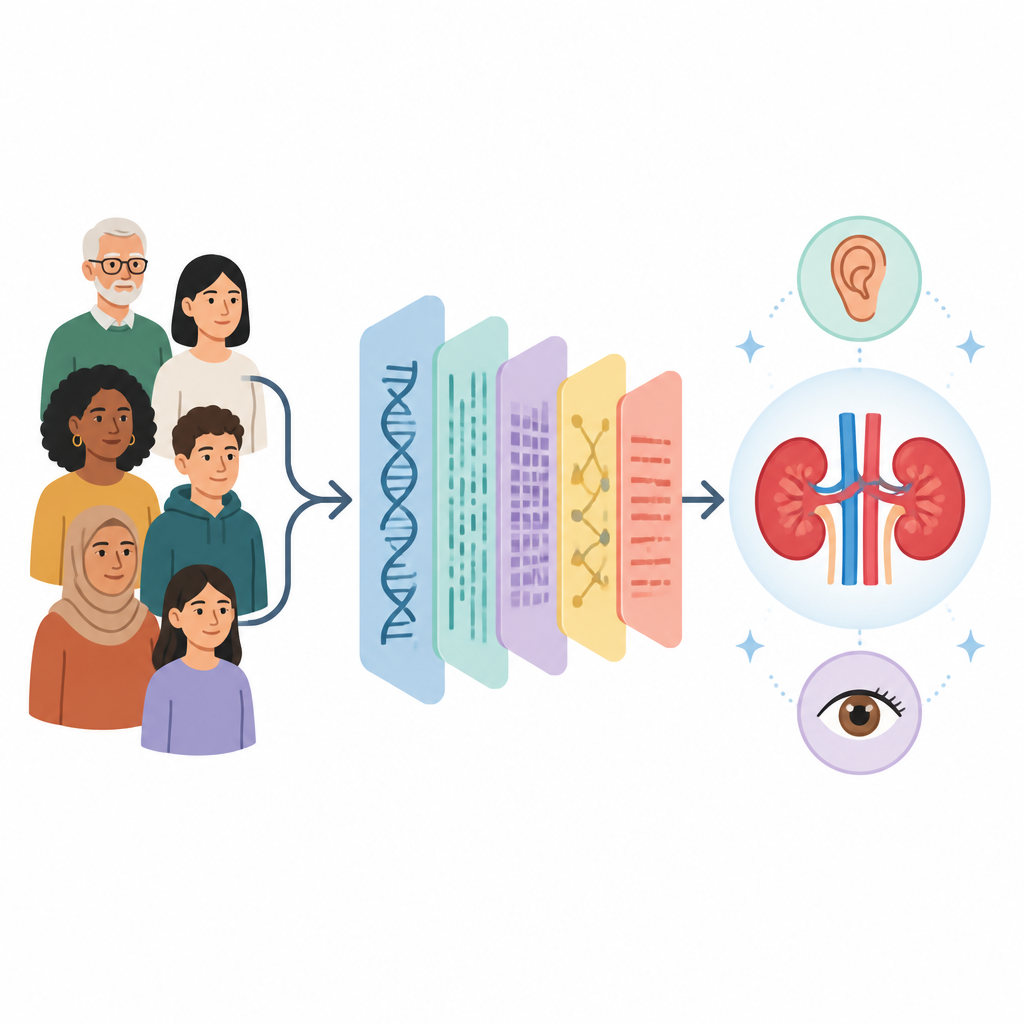
Le syndrome d’Alport est une maladie rénale héréditaire rare qui évolue souvent vers l’insuffisance rénale et peut aussi affecter l’audition et la vue. De nombreuses familles ne découvrent jamais la cause génétique précise, ce qui complique l’évaluation du risque familial et la planification des traitements et du suivi. Cette étude a appliqué plusieurs tests ADN et ARN de pointe à plus de 500 patients pour dresser un tableau beaucoup plus clair des altérations génétiques cachées à l’origine du syndrome d’Alport.
Une vue rapprochée d’un filtre rénal fragile
Le syndrome d’Alport endommage les filtres naturels du corps, en particulier dans les reins. Ces filtres reposent sur un réseau de protéines qui forment une fine couche de soutien dans les parois des vaisseaux sanguins. Trois gènes clés participent à la construction de ce réseau, et une altération de l’un d’eux peut affaiblir la structure. Quand cela se produit, du sang peut passer dans l’urine, des cicatrices peuvent s’accumuler dans les reins et, avec le temps, les filtres peuvent céder, parfois accompagnés de problèmes auditifs et oculaires qui partagent le même tissu de soutien.
Tests en couches pour trouver les mutations cachées
Les chercheurs ont étudié 555 personnes en Chine présentant un syndrome d’Alport confirmé par biopsie. Ils ont commencé par un test largement utilisé qui analyse les parties codantes des gènes, puis ont ajouté le séquençage du génome entier, le séquençage de l’ARN à partir de tissu rénal et le séquençage long reads pour les cas particulièrement énigmatiques. Cette approche progressive leur a permis de détecter non seulement des substitutions classiques d’une seule lettre d’ADN, mais aussi des altérations dans des régions non codantes, des pertes ou duplications d’segments d’ADN et des réarrangements plus complexes que les tests standards manquent souvent.
Une variété inattendue de modifications génétiques
Grâce à cette stratégie en couches, l’équipe a identifié des variants associés à la maladie chez plus de 91 % des patients et a répertorié 431 changements génétiques distincts, dont près de la moitié n’avaient jamais été signalés auparavant. La plupart des altérations se situaient dans les régions codantes des protéines, mais environ une sur six se trouvait dans des segments non codants de l’ADN qui influencent la façon dont les messages génétiques sont découpés et assemblés. Les scientifiques ont montré que certaines de ces modifications cachées entraînent l’insertion de segments supplémentaires dans le message ou l’omission complète de segments, perturbant la structure du réseau de collagène. Ils ont également mis au jour des formes jusque-là inconnues de grandes altérations d’ADN, notamment de longs fragments insérés à l’intérieur de lacunes géniques et des configurations complexes duplication–inversion qui modifient la production des messages géniques.
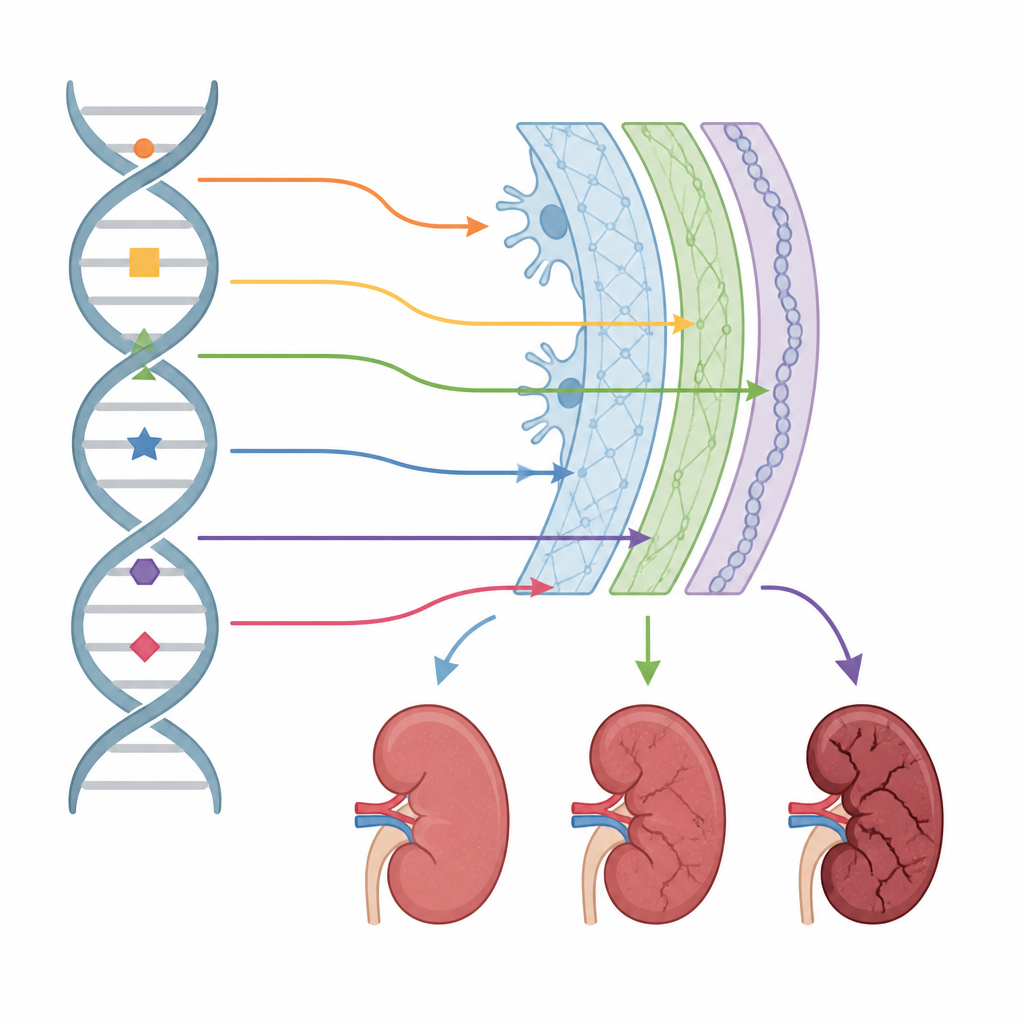
Corrélations entre modifications génétiques et symptômes
Étant donné que les patients disposaient de dossiers cliniques détaillés et d’échantillons de tissu rénal, les chercheurs ont pu relier des types spécifiques d’altérations de l’ADN à la manière dont la maladie se manifestait. Chez les hommes atteints de la forme liée à l’X, des mutations plus sévères étaient associées à une perte complète d’une chaîne de collagène clé lors des tests de coloration des reins et de la peau et à une progression plus rapide vers l’insuffisance rénale. Certains profils génétiques étaient liés à une probabilité accrue de perte auditive ou de kystes rénaux, et la perte auditive elle-même indiquait un risque plus élevé d’insuffisance rénale. En revanche, les personnes porteuses de variants plus bénins ou dont la maladie est héritée selon d’autres schémas avaient tendance à montrer une progression plus lente ou moins prévisible.
Ce que cela signifie pour les patients et les familles
Ce travail montre que se limiter aux segments géniques habituels peut manquer une part importante des altérations responsables du syndrome d’Alport. En combinant le séquençage du génome entier et le séquençage long reads avec des études d’ARN, les médecins peuvent établir un diagnostic génétique pour beaucoup plus de patients, clarifier les risques familiaux et mieux estimer qui peut être exposé à une dégradation rénale plus rapide ou à des complications supplémentaires. La même feuille de route de tests multi-couches peut être adaptée à d’autres maladies héréditaires, aidant à découvrir des mutations difficiles à détecter que les tests génétiques standards négligent.
Citation: Di, H., You, Z., Wang, L. et al. Sequential sequencing reveals the architecture and complexity of genomic variants in patients with Alport syndrome. Nat Commun 17, 4321 (2026). https://doi.org/10.1038/s41467-026-70936-9
Mots-clés: Syndrome d’Alport, génétique des maladies rénales, séquençage du génome entier, séquençage long reads, collagène IV