Clear Sky Science · fr
Extension de la microcinétique en champ moyen pour une modélisation précise et efficace des surfaces catalytiques hétérogènes complexes
Pourquoi les petites surfaces catalytiques comptent
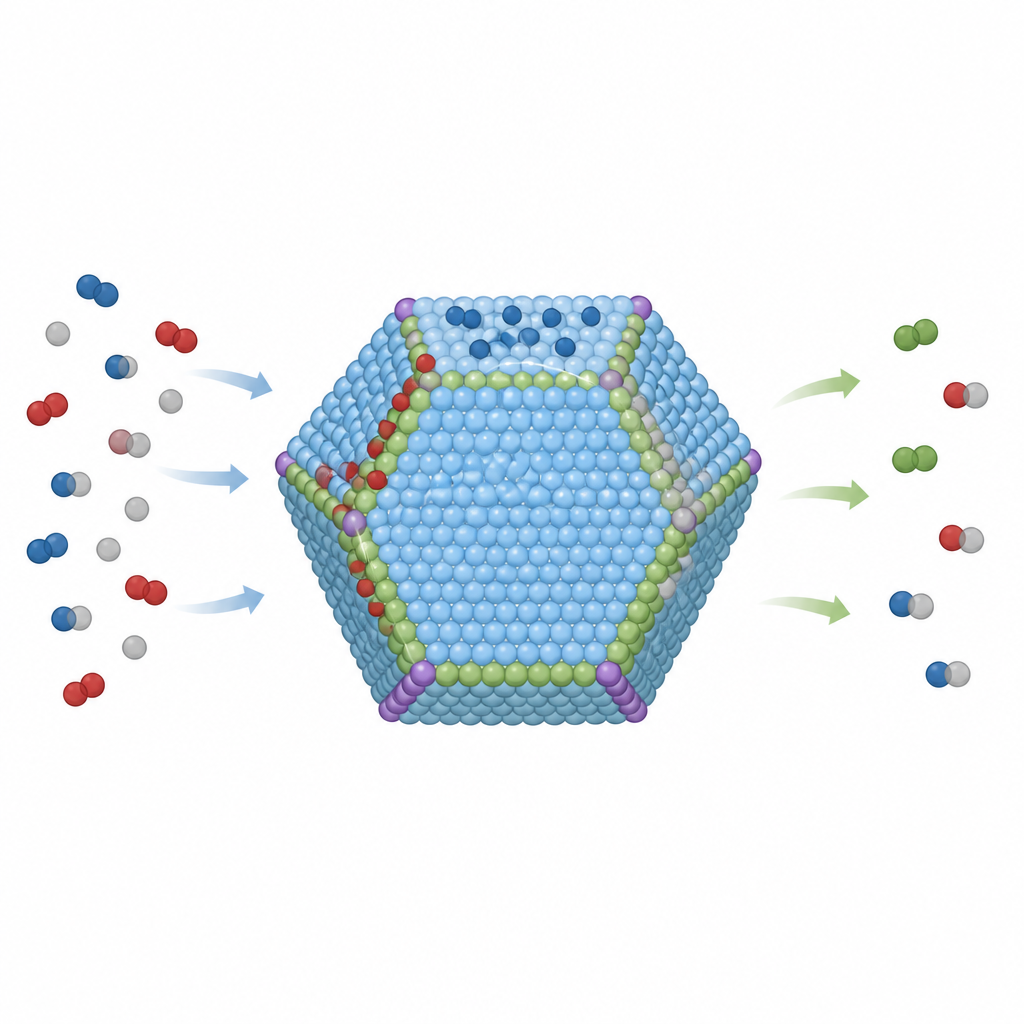
Les catalyseurs pilotent discrètement la plupart des réactions chimiques industrielles, de la production de carburants au traitement des gaz d’échappement. De nombreux catalyseurs modernes sont de minuscules particules métalliques dont la surface est un patchwork de quartiers atomiques différents. Pour concevoir de meilleurs catalyseurs par calcul, les scientifiques s’appuient sur des modèles mathématiques qui prédisent la vitesse des réactions. Cette étude propose une nouvelle façon de modéliser ces surfaces complexes plus fidèlement, sans supporter le coût informatique énorme des simulations entièrement détaillées.
Limites des modèles de raccourci actuels
Les approches de modélisation courantes traitent chaque face plane d’une particule métallique comme si elle fonctionnait isolément. Elles négligent souvent aussi l’encombrement de la surface quand de nombreuses molécules en réaction se disputent l’espace ou glissent d’un type de site à un autre. Ces raccourcis rendent les équations simples et rapides, mais peuvent donner des résultats trompeurs sur les caractéristiques de surface réellement actives et la vitesse de formation des produits. Des méthodes plus détaillées existent, qui suivent chaque événement sur une grille d’atomes, mais elles sont si coûteuses en calcul qu’elles ne permettent pas d’explorer un grand nombre de formes, tailles ou compositions de catalyseurs.
Une nouvelle façon de voir la particule entière
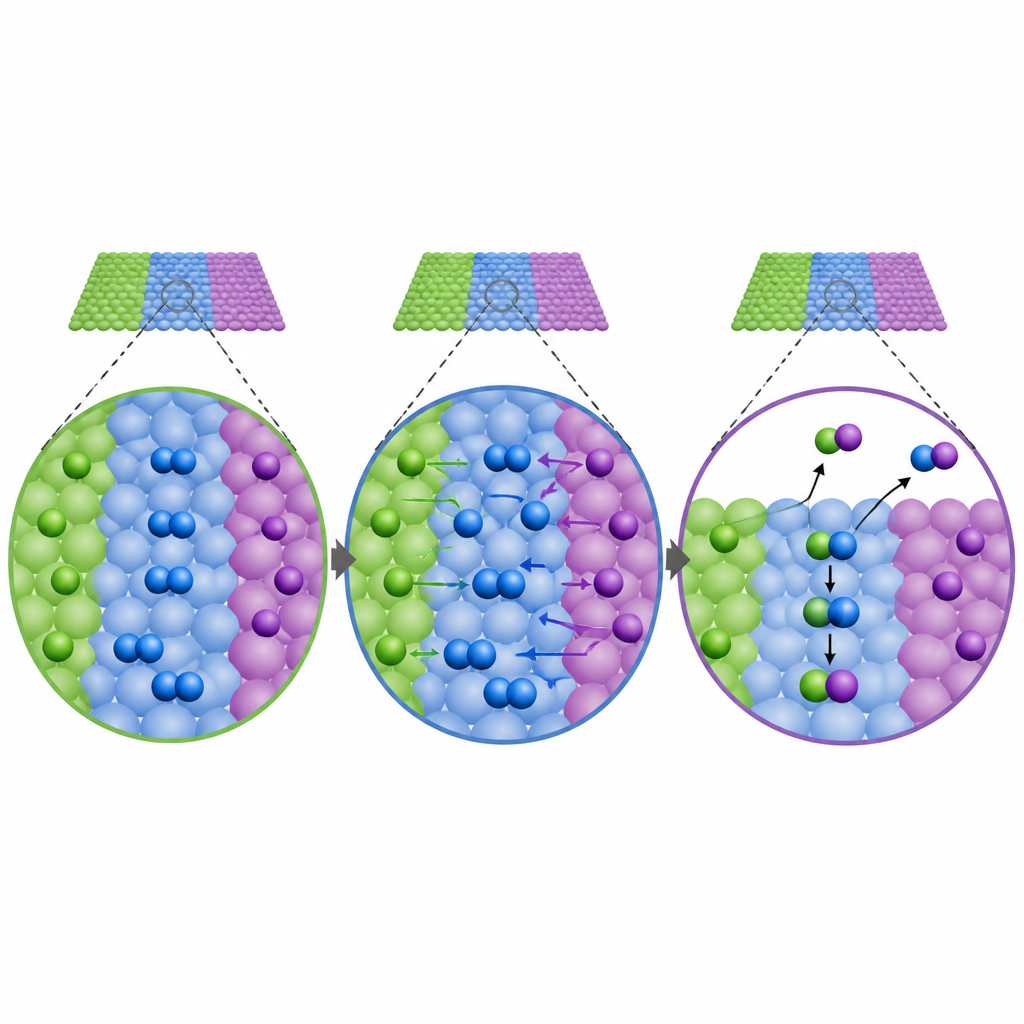
Les auteurs présentent un cadre étendu en champ moyen, appelé XMF, qui traite une nanoparticule métallique entière comme un système interagissant unique. Plutôt que d’isoler chaque facette de surface, la méthode relie différents types de sites par une description de la facilité de diffusion des molécules entre eux et de leur répulsion mutuelle lorsque la surface devient encombrée. Une idée clé est de se concentrer sur les espèces de surface les plus abondantes, souvent le monoxyde de carbone dans ce travail, et d’utiliser sa couverture moyenne pour ajuster les énergies réactionnelles sur toute la particule. En construisant un réseau d’équations compact basé sur ces informations, XMF suit comment les couvertures et les vitesses de réaction sur différents sites s’influencent mutuellement tout en restant aussi peu coûteux en calcul que les modèles simplifiés plus anciens.
Tester la méthode sur des réactions connues
Pour vérifier l’efficacité de la nouvelle approche, l’équipe a étudié la réaction eau-gaz décalée sur le platine, un processus bien connu qui aide à produire et purifier l’hydrogène. Ils ont comparé trois niveaux de la méthode XMF à des simulations cinétiques Monte Carlo détaillées, qui ont servi de référence numérique. Sur des surfaces planes de platine simples, XMF a reproduit fidèlement les vitesses de réaction, les énergies d’activation apparentes et l’étape élémentaire limitante, alors que les modèles en champ moyen standard échouaient lorsque la surface était fortement couverte de monoxyde de carbone. XMF a également rendu compte des changements de sites actifs préférentiels avec la température, saisissant les déplacements de l’étape limitante lorsque le système se réchauffe.
Quand les zones de surface coopèrent
La vraie force de XMF apparaît lorsque différentes zones de surface peuvent coopérer. Les auteurs ont construit un modèle simple combinant deux faces de platine de réactivités distinctes et ont autorisé la diffusion des molécules entre elles. Dans ce système couplé, le parcours réactionnel s’est réorganisé : l’eau se dissociait principalement sur des sites de type arête, tandis que des intermédiaires clés migraient vers des régions plus planes pour achever la réaction. XMF a capté la plus grande activité globale et la nouvelle étape limitante qui émergeait de cette coopération, alors que les modèles conventionnels additionnant simplement les contributions séparées de chaque face ne le pouvaient pas. Appliquée à des nanoparticules de platine réalistes de nombreuses tailles, l’étude a montré que les sites d’arête et de terrasse restent liés cinétiquement même pour des particules approchant centimètres, remettant en question l’idée que les grandes particules se comportent comme des lamelles indépendantes.
Vers des catalyseurs du monde réel
Les chercheurs ont enfin appliqué XMF à des systèmes plus complexes, incluant des nanoparticules de platine de formes variées et des alliages platine‑ruthénium proposés pour la production d’hydrogène à basse température. XMF a reproduit les tendances issues des simulations détaillées concernant les vitesses de réaction et les énergies d’activation apparentes, et a correctement identifié les compositions d’alliage prédites comme les plus actives, bien qu’il ait surestimé l’activité lorsque des molécules empoisonnantes se regroupaient étroitement sur plusieurs atomes adjacents de ruthénium. Malgré ces limites, le cadre permet une sélection rapide de milliers de structures candidates tout en tenant compte de l’encombrement de surface et de la communication entre sites.
Ce que cela signifie pour la découverte de catalyseurs
Pour un public non spécialiste, le message principal est que la structure fine d’une particule catalytique compte, et que coins, arêtes et régions planes ne fonctionnent pas en isolation. En offrant un outil de calcul capable de voir la particule entière tout en restant rapide, ce travail aide à combler l’écart entre la réalité atomique complexe et les calculs de conception pratiques. Le cadre XMF fournit des indications plus fiables sur les motifs de surface et les configurations d’alliage qui améliorent réellement les performances, facilitant des recherches plus rapides et mieux informées pour trouver de meilleurs catalyseurs pour de nombreuses réactions industrielles.
Citation: Wang, Y., Shen, T., Yang, Y. et al. Extending the mean-field microkinetics for an accurate and efficient modeling of complex heterogeneous catalyst surfaces. Nat Commun 17, 4426 (2026). https://doi.org/10.1038/s41467-026-70896-0
Mots-clés: catalyse hétérogène, modélisation microcinétique, catalyseurs nanoparticulaires, réaction eau-gaz décalée, alliages platine-ruthénium