Clear Sky Science · fr
Élargir le paysage génétique de la maladie du noyau poussiéreux : nouvelles variantes RYR1 chez des patients italiens
Quand les fibres musculaires perdent leur ordre habituel
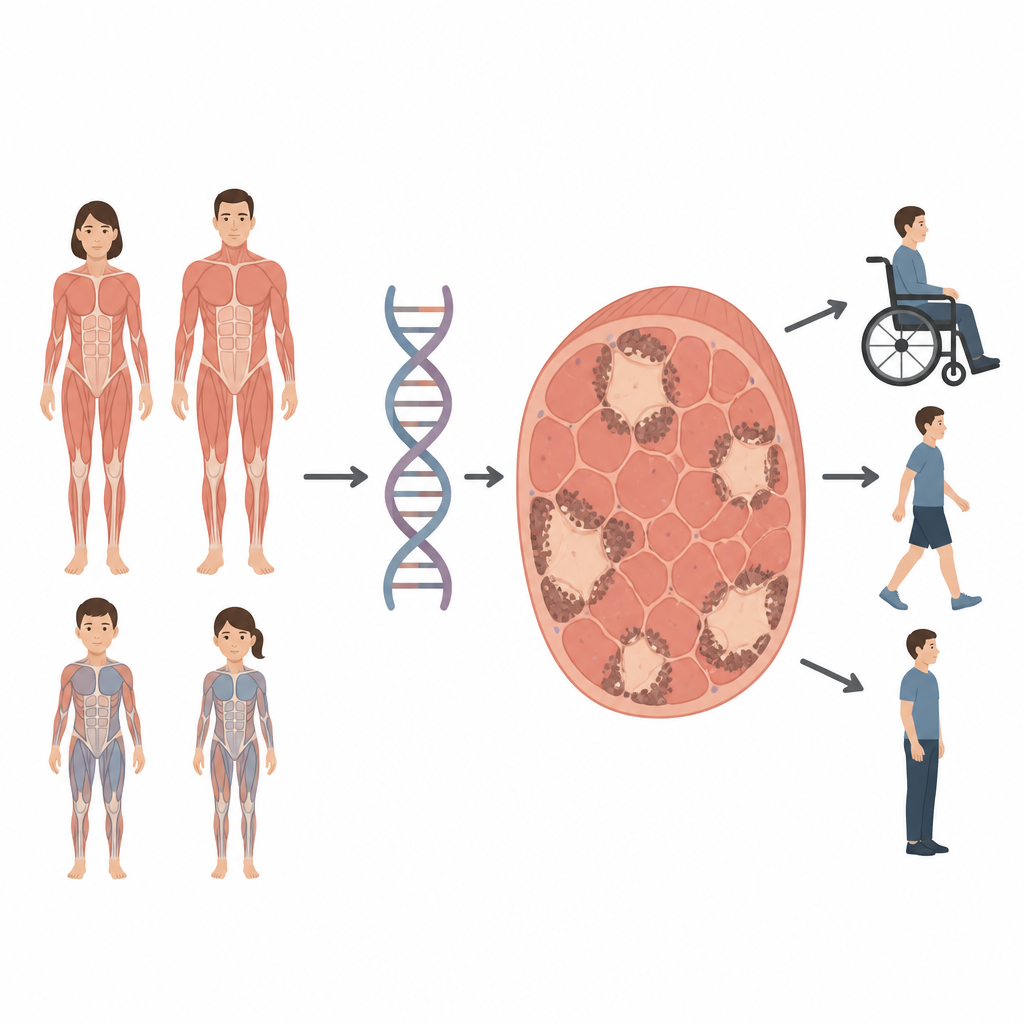
Certaines enfants et certains adultes développent une faiblesse musculaire qui rend la marche, la respiration ou même le maintien de la tête difficiles. Cette étude examine une affection musculaire rare appelée maladie du noyau poussiéreux, dans laquelle de petites zones à l’intérieur des cellules musculaires perdent leur structure normale. En retraçant comment les modifications d’un seul gène façonnent ce trouble, les chercheurs montrent qu’il peut toucher enfants et adultes et se transmettre de plusieurs façons, des informations importantes pour le diagnostic, le conseil génétique et les soins futurs.
Un regard rapproché sur un problème musculaire rare
Les myopathies à noyaux sont des maladies musculaires héréditaires où des portions des fibres musculaires, appelées noyaux, cessent de fonctionner correctement. La maladie du noyau poussiéreux est un sous-type récemment décrit, nommé d’après ses zones irrégulières et ponctuées remplies de matériel granulaire qui paraissent « poussiéreuses » au microscope. Jusqu’à présent, ce motif n’avait été observé que chez des personnes porteuses d’altérations délétères des deux copies d’un gène musculaire appelé RYR1 et qui présentaient en général une faiblesse d’apparition précoce. L’équipe italienne à l’origine de cette étude s’est donné pour objectif d’examiner quatre patients présentant le motif poussiéreux à la biopsie musculaire et de relier les observations cliniques, microscopiques et génétiques.
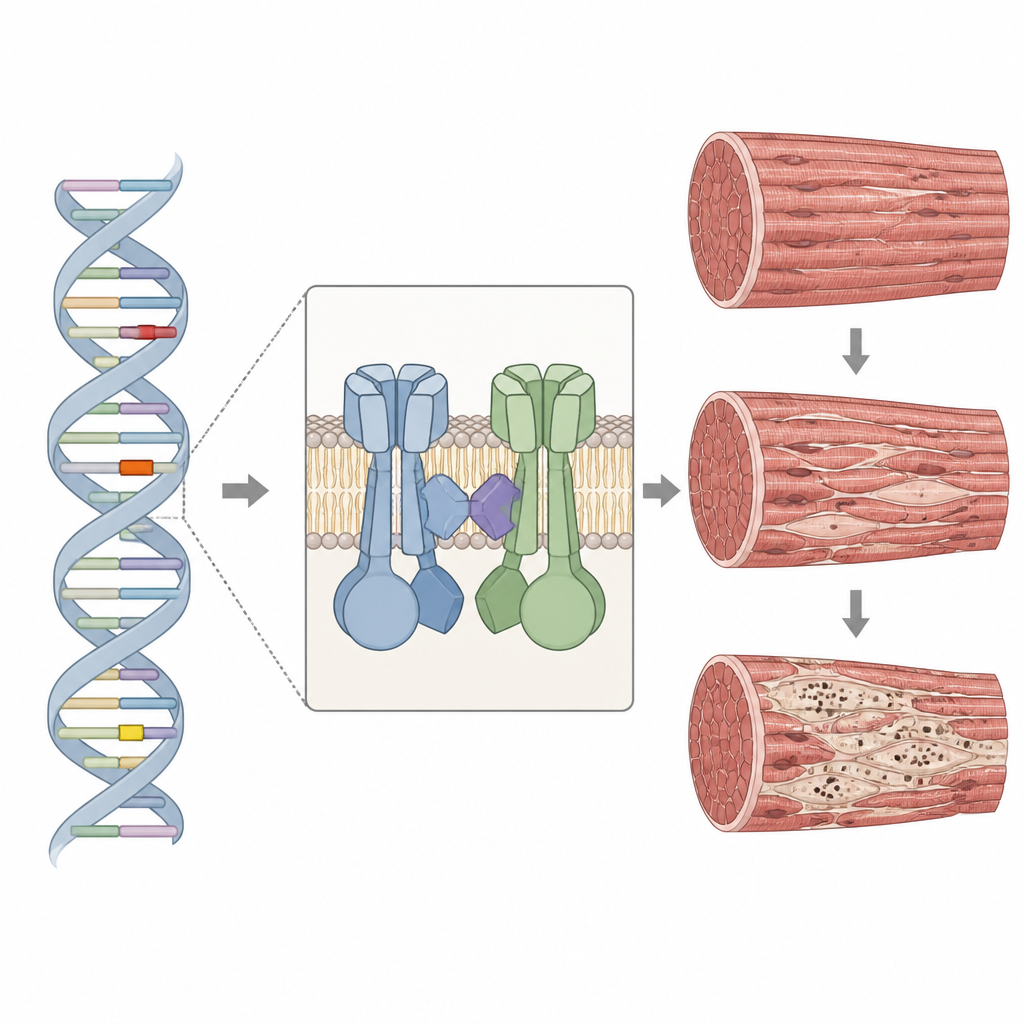
Comment la porte du calcium dans le muscle peut dysfonctionner
Le gène RYR1 fournit le plan d’un grand canal qui contrôle la libération du calcium à l’intérieur des cellules musculaires, une étape essentielle permettant la contraction. La protéine se situe dans une région spécialisée du muscle et travaille en étroite collaboration avec un canal partenaire qui capte les signaux électriques. Lorsque RYR1 est modifié, la libération de calcium peut être trop faible, trop importante ou mal coordonnée, ce qui affaiblit le muscle et perturbe son échafaudage interne. Chez les quatre patients, les chercheurs ont identifié des variantes rares dans des segments importants du gène RYR1, dont deux jamais décrites auparavant. Certaines altérations réduisaient la quantité de protéine RYR1 produite, tandis que d’autres modifiaient des points de contact critiques nécessaires au fonctionnement normal du canal.
Des patients de l’enfance à l’âge moyen
Les quatre personnes allaient du jeune enfant à l’adulte d’âge moyen et présentaient un spectre de sévérité. Deux filles avaient une faiblesse sévère d’apparition précoce avec des rétractions articulaires et une courbure de la colonne vertébrale ; l’une nécessitait un fauteuil roulant et une assistance respiratoire. Deux adultes, un homme et une femme, ont développé une faiblesse plus légère plus tard dans la vie, avec des difficultés comme monter des escaliers ou maintenir la tête, mais restaient capables de marcher. Une des femmes provenait d’une famille où plusieurs membres étaient atteints, ce qui indique qu’une seule copie altérée de RYR1 suffisait à provoquer la maladie. Cela contraste avec les deux cas pédiatriques, où soit les deux copies de RYR1 étaient modifiées, soit une altération entraînait la perte du produit de la copie affectée.
Ce que les prélèvements musculaires ont révélé
Les biopsies de muscles du bras ou de la cuisse montraient des fibres de tailles très variables et une augmentation du tissu conjonctif et adipeux, signes de dommage chronique. Des colorations spéciales mettaient en évidence les régions « poussiéreuses » caractéristiques : des zones irrégulières avec une activité enzymatique réduite et, dans certains échantillons, des granules violets-rouges à l’intérieur et autour des noyaux. Au microscope électronique à fort grossissement, ces noyaux se sont avérés être des zones où le motif strié normal des fibres musculaires se dégradait, les lignes Z étaient épaissies ou floues et les mitochondries faisaient défaut. Des marquages complémentaires ont montré que RYR1 et son canal partenaire formaient souvent des amas à l’intérieur et autour de ces noyaux, et que d’autres protéines structurelles s’y accumulaient aussi, suggérant une défaillance plus large de la machinerie reliant les signaux électriques à la contraction musculaire.
Pourquoi les modes de transmission importent
En combinant analyses génétiques, mesures protéiques et imagerie détaillée, les chercheurs ont conclu que le motif poussiéreux peut résulter d’altérations de RYR1 à la fois récessives et dominantes. Chez un adulte, une seule variante nouvelle dans une région d’interaction clé semblait suffisante pour perturber le canal calcique et son partenaire, tandis que chez un autre, deux variants situés sur la même copie du gène ont probablement modifié la sensibilité du canal. Ces observations montrent que la maladie du noyau poussiéreux fait partie d’un large ensemble de troubles musculaires liés à RYR1 qui peuvent apparaître à tout âge et suivre différentes voies de transmission. Pour les familles, cela signifie que la mise en évidence d’un motif poussiéreux à la biopsie doit déclencher des tests génétiques et un conseil attentifs ; pour les chercheurs, cela illustre comment de subtiles modifications d’une seule porte calcique peuvent remodeler le paysage intérieur du muscle humain.
Citation: Zanotti, S., Magri, F., Salani, S. et al. Expanding the genetic landscape of Dusty Core Disease: new RYR1 variants in Italian patients. Eur J Hum Genet 34, 609–618 (2026). https://doi.org/10.1038/s41431-026-02080-3
Mots-clés: Maladie du noyau poussiéreux, gène RYR1, myopathie congénitale, faiblesse musculaire, canal calcique