Clear Sky Science · fr
Atelier sur le développement thérapeutique pédiatrique des tumeurs rhabdoïdes
Pourquoi ce cancer infantile rare importe
Les tumeurs rhabdoïdes sont des cancers rares mais agressifs qui touchent le cerveau, les reins et les tissus mous de très jeunes enfants, souvent des nourrissons et des tout-petits. Les traitements actuels reposent sur des combinaisons de chirurgie, de chimiothérapie à haute dose et de radiothérapie, qui peuvent sauver certains enfants mais laissent beaucoup d’entre eux avec des séquelles durables et conduisent à des taux de survie faibles chez les patients les plus à risque. Cet article décrit comment des experts internationaux se sont réunis pour tracer une voie plus claire vers des médicaments plus intelligents et plus précis qui ciblent directement les vulnérabilités de ces tumeurs, visant à améliorer les taux de guérison tout en réduisant les dommages à long terme.
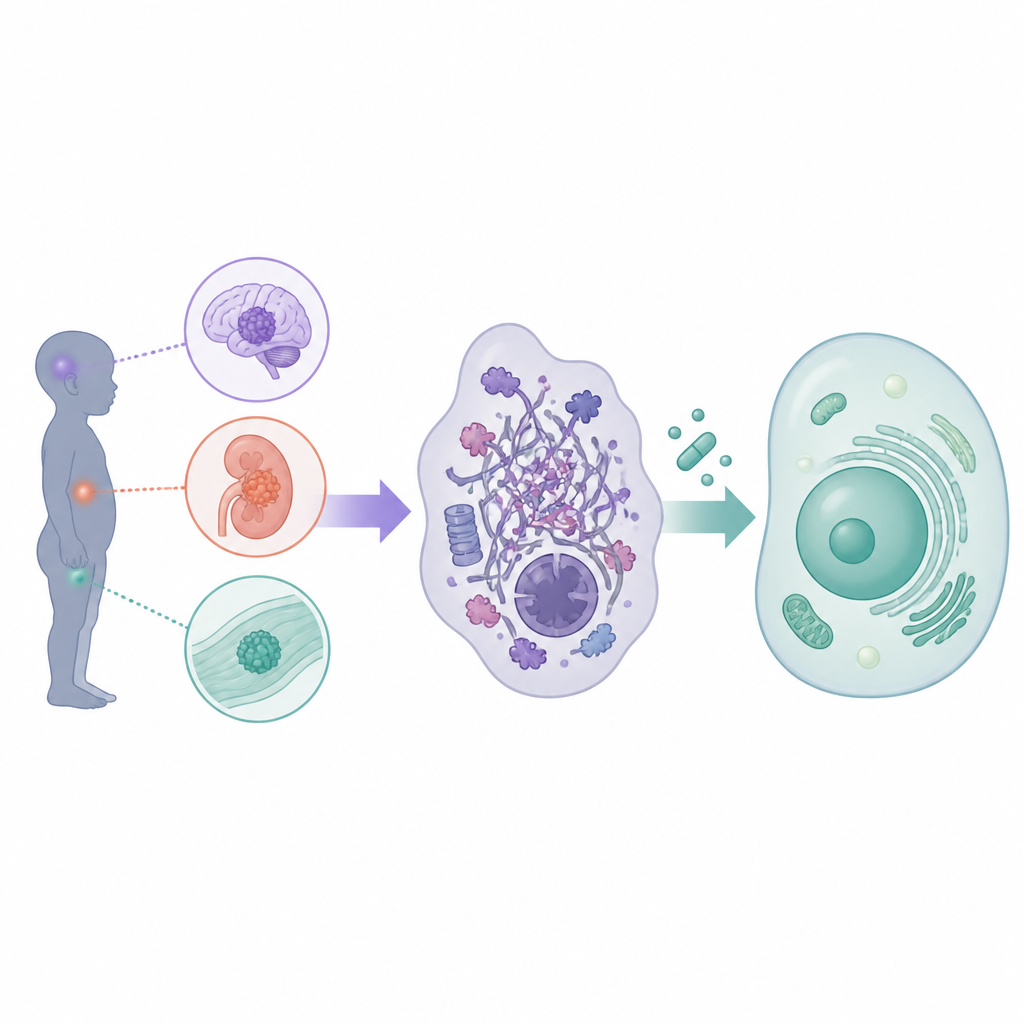
Qu’est‑ce que les tumeurs rhabdoïdes chez l’enfant
Les tumeurs rhabdoïdes se forment dans le système nerveux central, en particulier chez les très jeunes enfants où elles sont appelées tumeurs tératoïdes atypiques ou rhabdoïdes, et en dehors du cerveau dans les reins, le foie et d’autres tissus mous. Environ 200 enfants par an seulement sont diagnostiqués en Europe et aux États‑Unis, pourtant la maladie est souvent rapidement fatale. La thérapie standard consiste en une chirurgie maximale compatible avec la sécurité, suivie d’une chimiothérapie intensive, fréquemment combinée à la radiothérapie et parfois à une greffe de cellules souches. Ces approches ont porté la survie autour de 30 à 40 pour cent dans de nombreux cas, mais au prix de toxicités graves à court et à long terme, incluant des dommages au cerveau en développement. Parce que chaque essai n’a enrôlé qu’un petit nombre de patients et s’est souvent concentré soit sur les tumeurs cérébrales soit sur les tumeurs corporelles, il est nécessaire de coordonner les efforts et de repenser la façon dont les nouveaux médicaments sont amenés en clinique.
Un moteur commun derrière différents types de tumeurs
Bien que les tumeurs rhabdoïdes puissent apparaître dans différents organes, les chercheurs ont constaté qu’elles partagent presque toutes le même défaut central dans leur machinerie cellulaire. Dans les cellules saines, un groupe de protéines connu sous le nom de complex SWI/SNF aide à organiser l’ADN et à contrôler quels gènes sont activés ou désactivés. Dans les tumeurs rhabdoïdes, des éléments clés de ce complexe, le plus souvent la protéine SMARCB1 et plus rarement SMARCA4, sont inactivés. Cela n’entraîne pas une avalanche de mutations ; en revanche, cela reconfigure subtilement le contrôle de l’activité génétique, immobilise les cellules dans un état immature et favorise une croissance incontrôlée. Parce que ce défaut central est partagé entre tumeurs cérébrales et tumeurs corporelles, l’atelier a conclu que de nombreux médicaments ciblés pourraient être conçus et testés pour l’ensemble des tumeurs rhabdoïdes, à condition que les agents destinés aux tumeurs cérébrales puissent franchir la barrière hémato‑encéphalique.
Nouvelles vulnérabilités pour des médicaments précis
Les chercheurs ont commencé à exploiter le câblage inhabituel des tumeurs rhabdoïdes pour révéler leurs principales vulnérabilités. L’article passe en revue des dizaines de cibles candidates puis met en avant une courte liste convenue lors de l’atelier. Une priorité majeure est DCAF5, une protéine qui reconnaît le complexe SWI/SNF endommagé et le marque pour destruction ; bloquer DCAF5 dans des modèles de laboratoire permet à assez de complexe de se reformer et ralentit fortement la croissance tumorale tout en épargnant les cellules normales. Une autre cible clé est MDM2, un régulateur de la célèbre protéine gardienne p53. Parce que les cellules rhabdoïdes conservent un système p53 largement intact mais maintenu réprimé, des médicaments inhibant MDM2 peuvent relâcher ce frein naturel et réduire la taille des tumeurs chez la souris. Le groupe a aussi discuté des moyens d’attaquer le complexe partenaire PRC2, en particulier en dégradant sa sous‑unité EZH2 plutôt qu’en se contentant d’inhiber son activité enzymatique, ce qui pourrait produire des effets plus profonds et durables que la génération de médicaments initiale telle que le tazémétostat.
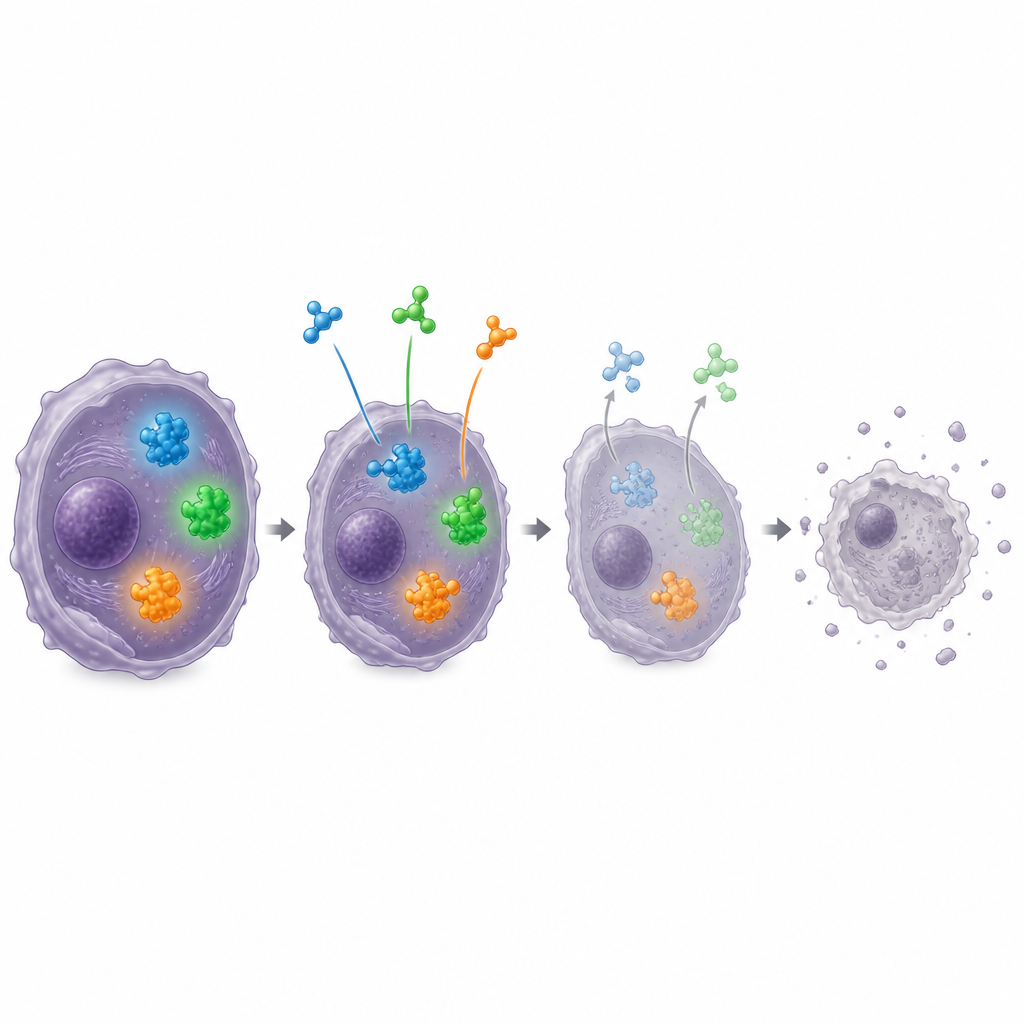
Combiner les forces pour un meilleur traitement
Compte tenu de la complexité de la biologie tumorale, l’atelier a mis l’accent sur les associations d’agents susceptibles de fonctionner mieux en synergie que séparément. Les données de laboratoire suggèrent que l’association d’agents ciblant EZH2 avec des inhibiteurs de MDM2, ou avec des inhibiteurs sélectifs de l’export nucléaire qui aident des protéines suppresseures de tumeurs mal localisées à revenir dans le noyau, peut être particulièrement efficace. D’autres partenaires prometteurs incluent des médicaments contre les kinases Aurora, EED, ou des protéines de la réponse aux dommages de l’ADN telles qu’ATR, qui peuvent aider à surmonter la résistance aux agents uniques. Les experts ont aussi envisagé des médicaments bloquant les récepteurs des facteurs de croissance FGFR et PDGFR dans les sous‑groupes où ces signaux sont hyperactifs, ainsi que des approches immunitaires telles que les inhibiteurs de points de contrôle et les lymphocytes T modifiés pour les tumeurs riches en infiltration immunitaire.
Collaboration mondiale pour aider les enfants concernés
L’article conclut que les enfants atteints de tumeurs rhabdoïdes ont un besoin urgent de traitements à la fois plus efficaces et moins toxiques que les combinaisons agressives actuelles de chimiothérapie et de radiothérapie. Parce que ces cancers partagent un défaut sous‑jacent commun dans leur machinerie de contrôle des gènes, les auteurs soutiennent que des stratégies ciblées peuvent être développées pour les tumeurs cérébrales et corporelles selon les mêmes principes de base. L’atelier a créé un consortium international transatlantique pour tester les cibles et les paires de médicaments les plus prometteuses dans des modèles précliniques rigoureux, en suivant des standards communs afin que les résultats puissent être comparés et combinés. Leur objectif à long terme est de traduire ces résultats en essais cliniques de phase précoce bien conçus et, finalement, en plans de traitement mondiaux coordonnés, offrant aux familles une meilleure chance de guérison avec moins d’effets secondaires à vie.
Citation: Montiel Equihua, C., Molenaar, J.J., Areso, I. et al. Paediatric Therapeutic Development Workshop on rhabdoid tumours. Br J Cancer 134, 1510–1528 (2026). https://doi.org/10.1038/s41416-026-03348-7
Mots-clés: tumeurs rhabdoïdes, cancer cérébral de l’enfant, thérapie ciblée, inhibiteurs d’EZH2, inhibiteurs de MDM2