Clear Sky Science · en
Paediatric Therapeutic Development Workshop on rhabdoid tumours
Why this rare childhood cancer matters
Rhabdoid tumours are rare but aggressive cancers that strike the brains, kidneys and soft tissues of very young children, often babies and toddlers. Current treatments rely on combinations of surgery, high dose chemotherapy and radiotherapy, which can save some lives but leave many children with lasting side effects and still result in low survival rates for those at highest risk. This article describes how international experts came together to chart a clearer path toward smarter, more precise medicines that directly target the weak spots of these tumours, aiming to improve cure rates while reducing long term harm.
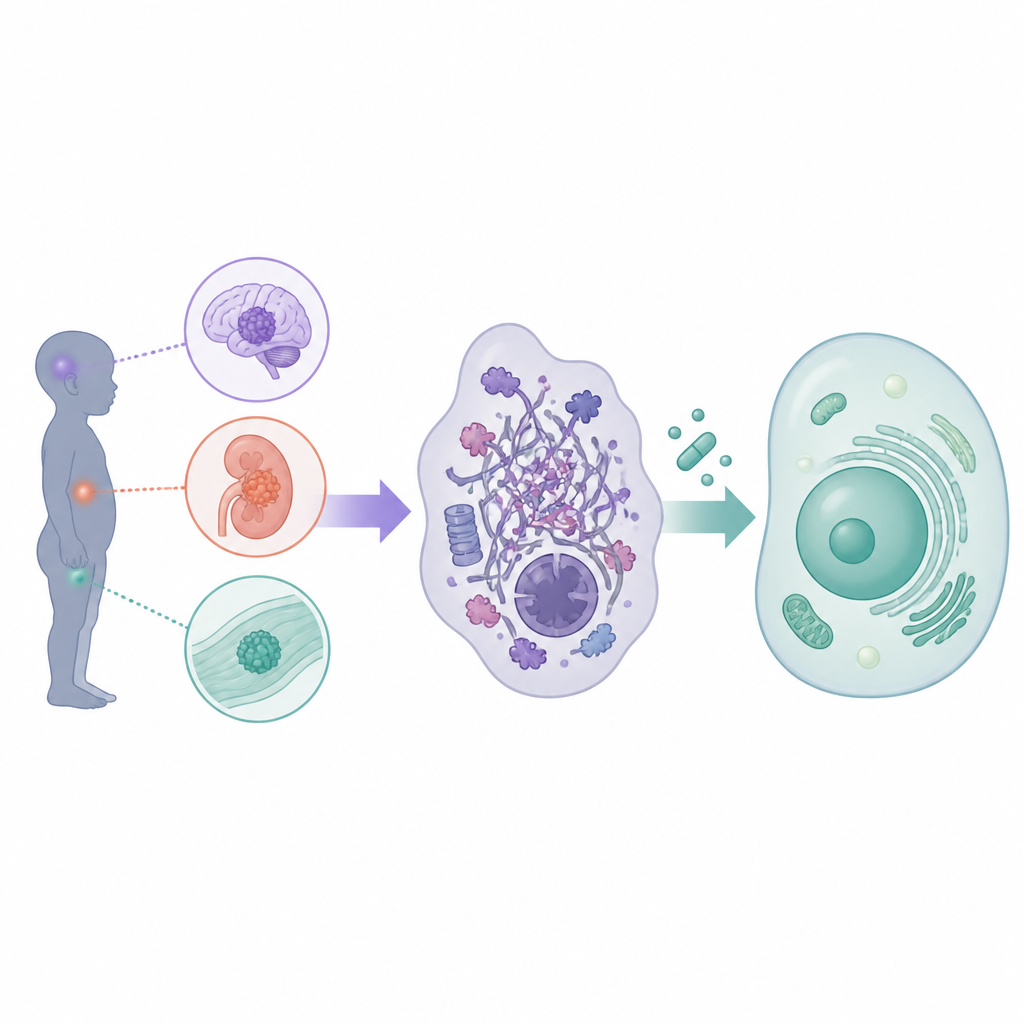
What are rhabdoid tumours in children
Rhabdoid tumours form in the central nervous system, especially in very young children where they are called atypical teratoid or rhabdoid tumours, and outside the brain in the kidneys, liver and other soft tissues. Only about 200 children a year are diagnosed across Europe and the United States, yet the disease is often rapidly fatal. Standard therapy involves maximal safe surgery followed by intensive chemotherapy, frequently combined with radiotherapy and sometimes stem cell rescue. These approaches have pushed survival to around 30 to 40 percent in many cases, but at the cost of serious short and long term toxicities, including damage to the developing brain. Because each trial has enrolled only small numbers of patients and has often focused on either brain or body tumours separately, there is a strong need to coordinate efforts and rethink how new drugs are brought to the clinic.
A common engine behind different tumour types
Although rhabdoid tumours can arise in different organs, scientists have learned that they almost all share the same core defect in their cellular machinery. In healthy cells, a group of proteins known as the SWI slash SNF complex helps to organise DNA and control which genes are switched on or off. In rhabdoid tumours, key parts of this complex, most often the protein SMARCB1 and less commonly SMARCA4, are disabled. This does not flood the cell with mutations; instead, it subtly rewires the control of gene activity, freezing cells in an immature state and promoting uncontrolled growth. Because this core glitch is shared across brain and body tumours, the workshop concluded that many targeted drugs could be designed and tested for all rhabdoid tumours together, provided that agents for brain tumours can cross the blood brain barrier.
New weak spots for precise drugs
Researchers have begun to exploit the unusual wiring of rhabdoid tumours to uncover their main vulnerabilities. The article reviews dozens of candidate targets and then highlights a short list agreed at the workshop. One top priority is DCAF5, a protein that recognises the damaged SWI slash SNF complex and tags it for destruction; blocking DCAF5 in laboratory models allows enough of the complex to reform and sharply slows tumour growth while sparing normal cells. Another key target is MDM2, a regulator of the well known guardian protein p53. Because rhabdoid cells still carry a largely intact p53 system but keep it suppressed, drugs that inhibit MDM2 can release this natural brake and shrink tumours in mice. The group also discussed ways to attack the partner complex PRC2, especially by degrading its subunit EZH2 rather than merely inhibiting its enzyme activity, which may give deeper and more lasting effects than the first generation drug tazemetostat.
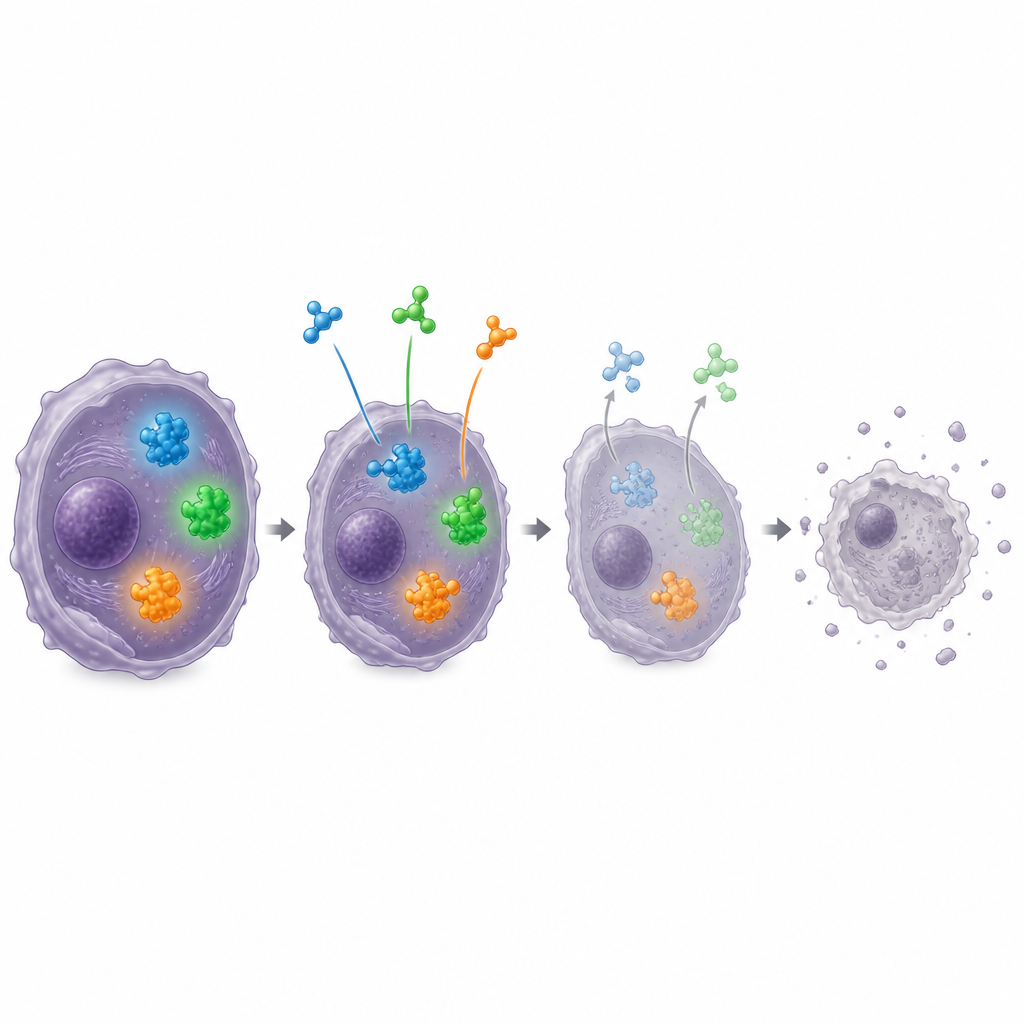
Combining forces for better treatment
Given the complexity of tumour biology, the workshop focused strongly on combinations of agents that might work better together than alone. Laboratory data suggest that pairing EZH2 targeting drugs with MDM2 inhibitors, or with selective inhibitors of nuclear export that help mislocated tumour suppressor proteins return to the cell nucleus, can be particularly effective. Other promising partners include drugs against aurora kinases, EED, or DNA damage response proteins such as ATR, which may help overcome resistance to single agents. The experts also considered drugs that block growth factor receptors FGFR and PDGFR in subgroups where these signals are overactive, and immune approaches such as checkpoint inhibitors and engineered T cells for tumours rich in immune cell infiltration.
Global teamwork to help affected children
The article concludes that children with rhabdoid tumours urgently need treatments that are both more effective and less toxic than today’s aggressive combinations of chemotherapy and radiotherapy. Because these cancers share a common underlying fault in their gene control machinery, the authors argue that targeted strategies can be developed for both brain and body tumours using the same basic principles. The workshop created an international, trans Atlantic consortium to test the most promising targets and drug pairs in rigorous preclinical models, following agreed standards so results can be compared and combined. Their long term goal is to translate these findings into well designed early phase clinical trials and ultimately into coordinated global treatment plans, offering families a better chance of cure with fewer lifelong side effects.
Citation: Montiel Equihua, C., Molenaar, J.J., Areso, I. et al. Paediatric Therapeutic Development Workshop on rhabdoid tumours. Br J Cancer 134, 1510–1528 (2026). https://doi.org/10.1038/s41416-026-03348-7
Keywords: rhabdoid tumours, childhood brain cancer, targeted therapy, EZH2 inhibitors, MDM2 inhibitors