Clear Sky Science · fr
Zinc finger BED-type contenant 6 (ZBED6) atténue la fibrose cardiaque en inhibant la transcription de Piezo1 et la translocation nucléaire de YAP
Pourquoi la cicatrisation cardiaque compte
Après un infarctus, l’organisme se précipite pour réparer le muscle endommagé. Mais au lieu de reconstruire un tissu solide et battant, le cœur forme souvent une cicatrice rigide. Cette cicatrisation, appelée fibrose cardiaque, rend la pompe cardiaque moins efficace et peut engager le patient sur la voie de l’insuffisance cardiaque. L’étude présentée ici met au jour un facteur protecteur intrinsèque dans les cellules de soutien du cœur capable de réduire la formation de cicatrice, et décrit la chaîne moléculaire qui active ou désactive cette protection.
Un acteur caché de la réparation cardiaque
La plupart des recherches sur les maladies cardiaques se concentrent sur les cardiomyocytes — les cellules qui se contractent et expulsent le sang. Pourtant, le cœur contient aussi de nombreux fibroblastes, des cellules de soutien qui tissent la trame structurelle entre les fibres musculaires. Lors d’une lésion, ces fibroblastes se transforment en une forme très active appelée myofibroblastes, produisant du collagène et d’autres fibres qui constituent le tissu cicatriciel. Les auteurs se sont intéressés à une protéine peu connue, ZBED6, un interrupteur génétique présent seulement chez les mammifères placentaires, pour savoir si elle régule l’intensité de la réponse des fibroblastes après un infarctus.
Trop peu de protection, trop de cicatrice
Chez des souris ayant subi des infarctus expérimentaux, les chercheurs ont mesuré les niveaux de ZBED6 dans le cœur en cours de cicatrisation. Ils ont constaté que ZBED6 chutait fortement dans les cœurs lésés et dans des fibroblastes cardiaques de souris en culture activés par la molécule pro‑cicatrisation TGF‑β. Ils ont ensuite modifié génétiquement des souris pour qu’elles produisent un excédent de ZBED6 spécifiquement dans les fibroblastes cardiaques. Chez ces animaux, la fonction cardiaque après infarctus était mieux préservée : les cavités de pompage restaient proches des dimensions normales, la fraction d’éjection s’améliorait et la surface de cicatrice était réduite. Des marqueurs d’activité fibrotique — comme le collagène et la protéine contractile α‑SMA — diminuaient dans le tissu cardiaque. À l’inverse, lorsque l’équipe a utilisé un virus pour diminuer ZBED6 uniquement dans les fibroblastes, les animaux ont développé une aggravation de la dilatation cardiaque, une baisse de la fonction d’éjection, davantage de collagène et un profil d’expression génique fibrotique amplifié — et ce sans déclencheur de lésion supplémentaire.
Comment ZBED6 freine un capteur de force
En approfondissant leur analyse, les auteurs ont cherché les gènes que ZBED6 pourrait contrôler et ont ciblé Piezo1, un gros canal ionique qui détecte les forces mécaniques telles que l’étirement et la pression. Piezo1 a été impliqué dans la façon dont les cellules perçoivent la rigidité et dans le remodelage cardiaque délétère. Ici, l’équipe montre que les niveaux de Piezo1 augmentent dans les cœurs endommagés et dans les fibroblastes activés, mais diminuent lorsque ZBED6 est sur‑exprimé et augmentent encore lorsque ZBED6 est silencé. Par des essais biochimiques, ils démontrent que ZBED6 se lie directement à des sites spécifiques de la région promotrice du gène Piezo1 et agit comme un frein sur son activité. Lorsque ces sites de liaison sont mutés, ou lorsque ZBED6 est réduit, Piezo1 devient plus actif.
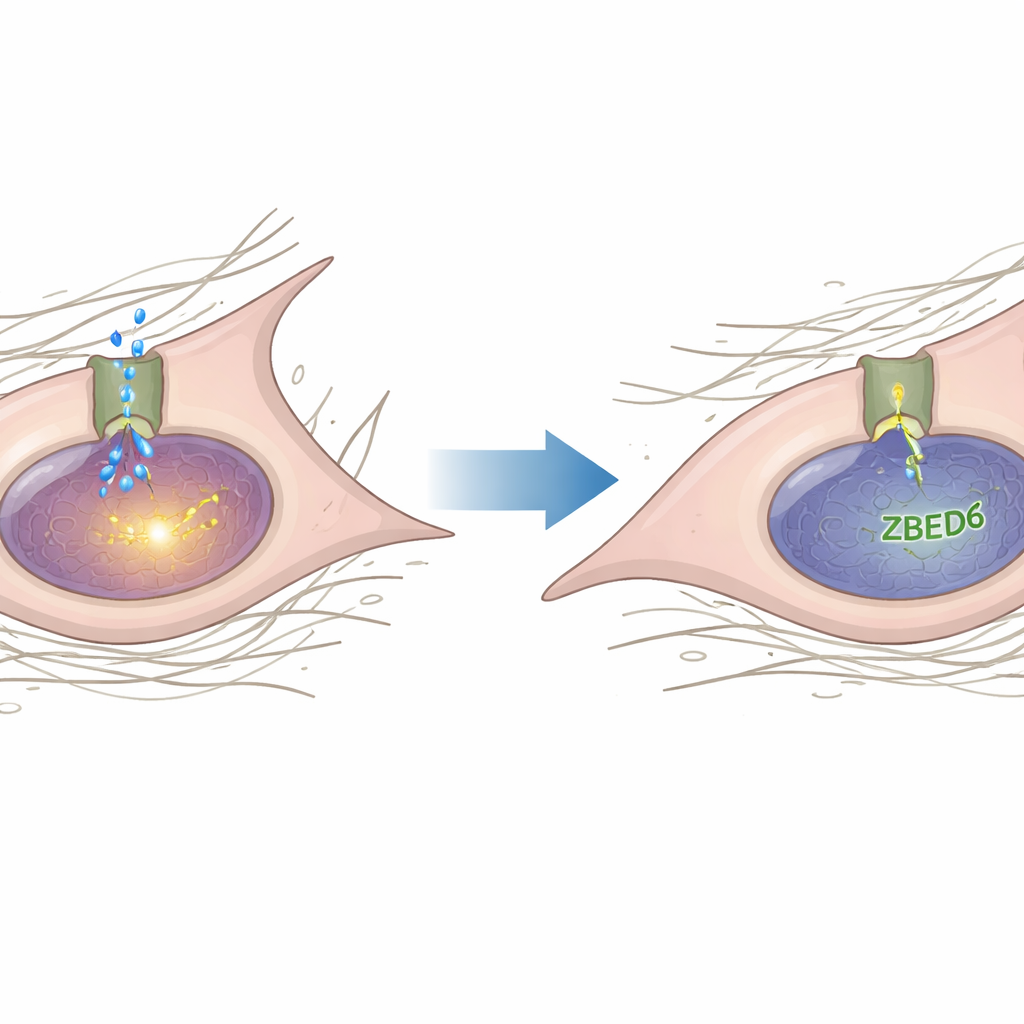
Du capteur de force aux signaux pro‑cicatrisants
L’histoire ne s’arrête pas à Piezo1. Ce canal, lorsqu’il est ouvert, laisse entrer des ions dans les fibroblastes et favorise la translocation d’une autre protéine, YAP, du cytoplasme vers le noyau. À l’intérieur du noyau, YAP coopère avec d’autres facteurs pour activer des gènes qui stimulent la croissance cellulaire, la migration et la production de fibres. Les chercheurs ont montré que l’activation de Piezo1 par un agoniste chimique augmentait la prolifération des fibroblastes, leur mobilité et l’expression des gènes fibrotique, et favorisait l’entrée de YAP dans le noyau. Le blocage de Piezo1, soit par un inhibiteur ciblé soit par la réduction de son expression, diminuait la présence nucléaire de YAP. Fait important, l’activation de Piezo1 pouvait annuler l’effet apaisant d’un excès de ZBED6 sur les fibroblastes, indiquant que Piezo1 se situe entre ZBED6 et YAP dans cette cascade de signalisation.
Ce que cela pourrait signifier pour de futurs traitements
Globalement, les résultats décrivent une logique simple : lorsque les niveaux de ZBED6 sont élevés dans les fibroblastes cardiaques, ils maintiennent le capteur de force Piezo1 sous contrôle, ce qui limite l’entrée de YAP dans le noyau et tempère le programme de cicatrisation. Quand ZBED6 est perdu ou réduit, Piezo1 devient hyperactif, YAP envahit le noyau et les fibroblastes déposent un excès de tissu cicatriciel. Pour les patients, cela suggère une nouvelle approche pour la récupération après infarctus. Plutôt que de se focaliser uniquement sur le sauvetage du muscle en train de mourir, des thérapies pourraient viser à augmenter l’activité de ZBED6 ou à atténuer l’axe Piezo1–YAP dans les fibroblastes, orientant la réparation vers un tissu cardiaque plus souple et moins cicatriciel.
Citation: Wu, H., Jiang, Wt., Zhao, Qy. et al. Zinc finger BED-type containing 6 (ZBED6) ameliorates cardiac fibrosis by inhibiting Piezo1 transcription and YAP nuclear translocation. Acta Pharmacol Sin 47, 1162–1175 (2026). https://doi.org/10.1038/s41401-025-01717-1
Mots-clés: fibrose cardiaque, insuffisance cardiaque, canaux ioniques mécanosensibles, fibroblastes, transduction du signal