Clear Sky Science · fr
L’hypoxie induit le microARN-27b responsable de l’endoreplication pathologique cardiaque dans les maladies du cœur
Quand les cellules cardiaques grandissent de la mauvaise façon
La maladie cardiaque ne se résume pas aux artères bouchées ou à une pompe défaillante. Au cœur du muscle cardiaque, des cellules individuelles peuvent commencer à recopier leur ADN sans se diviser, devenant surdimensionnées et remplies de noyaux supplémentaires. Ce travail explore pourquoi cela se produit en conditions de faible oxygène et révèle un interrupteur moléculaire caché qui relie la pénurie d’oxygène, la perturbation de la production d’énergie et la croissance cellulaire cardiaque anormale, pointant vers des stratégies thérapeutiques utilisant des outils déjà disponibles en clinique.
Faible oxygène et cœurs stressés
Lorsque le cœur doit pousser le sang contre une pression élevée, comme dans l’hypertension artérielle chronique ou une valve rétrécie, sa chambre principale s’épaissit pour compenser. À mesure que la paroi musculaire s’épaissit, de petites zones reçoivent moins de sang frais et deviennent pauvres en oxygène. Dans ces poches, une protéine capteur à l’intérieur des cellules, appelée HIF1α, s’active et remodèle l’utilisation des carburants par les cardiomyocytes. Les auteurs ont cherché à comprendre comment ce détecteur d’oxygène se relie à l’habitude étrange des cellules cardiaques de recopier leur ADN sans se diviser, ce qui conduit à des cellules excessivement grandes, multinucléées et, finalement, à une diminution de l’efficacité de la pompe cardiaque.
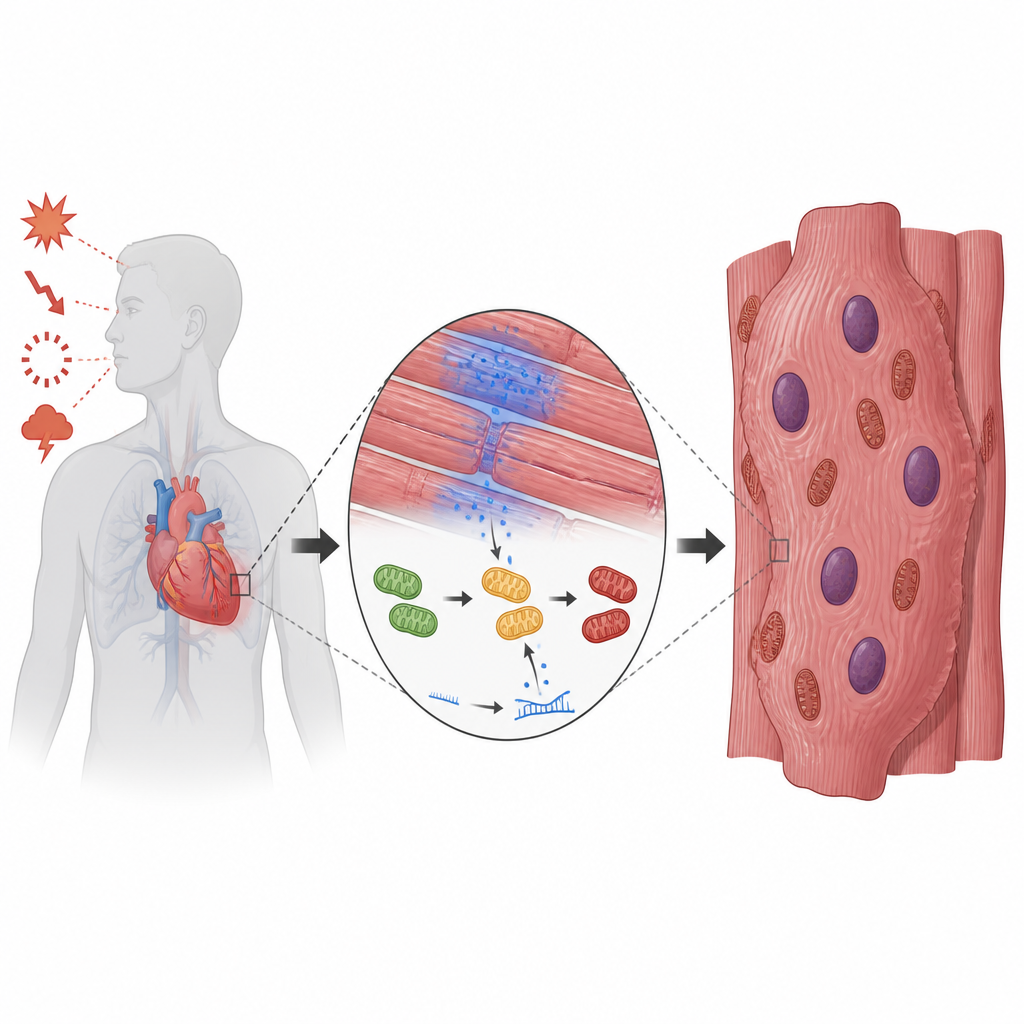
Un petit ARN qui contrôle l’énergie
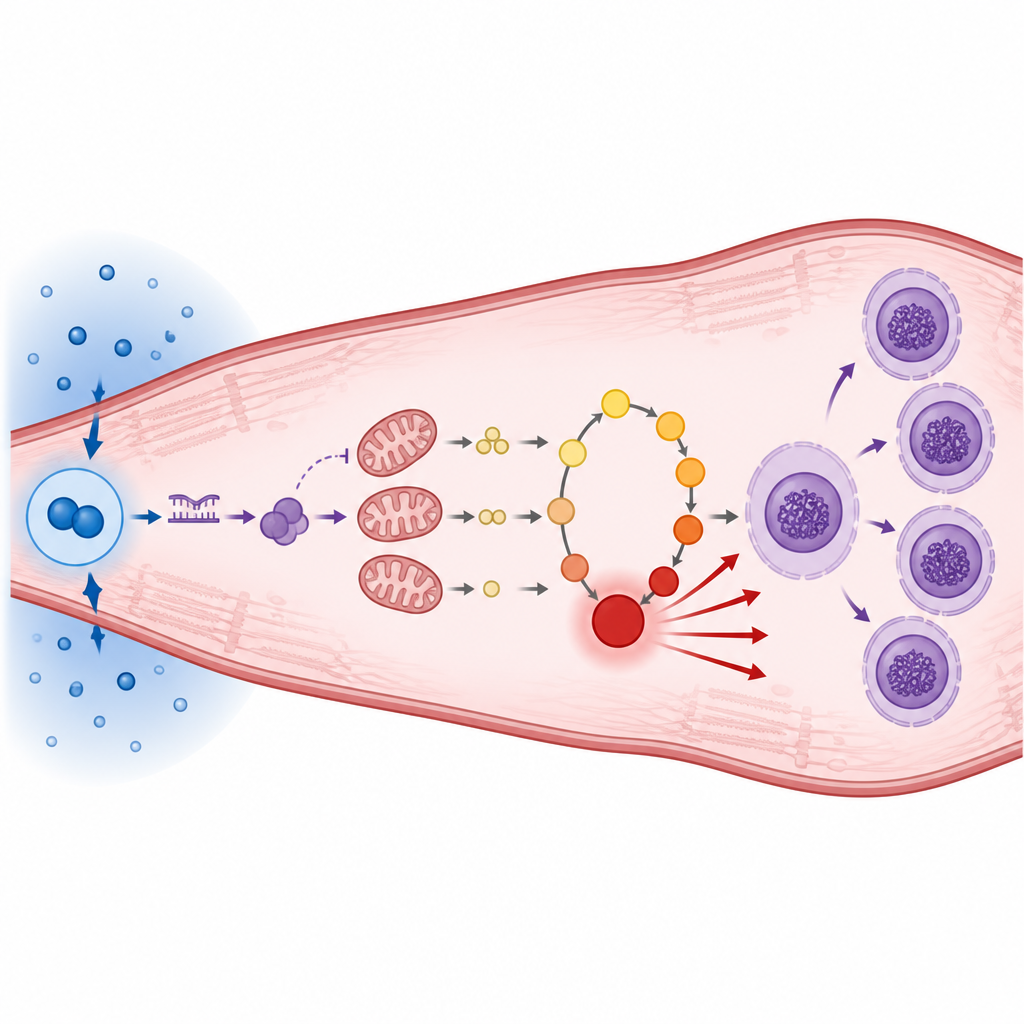
En comparant des modèles murins de surcharge cardiaque avec des biopsies de cœurs humains, l’équipe a observé un schéma inverse : lorsque les niveaux de HIF1α étaient élevés, une composante clé de la centrale cellulaire, ATP5A1, était basse, et les réserves énergétiques globales diminuaient. Plutôt que d’agir directement sur cette protéine de production d’énergie, HIF1α active un très petit fragment d’acide nucléique appelé microARN-27b-5p. Ce microARN fonctionne comme un variateur fin, se liant au message codant ATP5A1 et bloquant sa production. À la chute d’ATP5A1, les petites turbines des mitochondries tournent moins efficacement, la production d’ATP chute et une molécule associée, l’ADP, s’accumule à l’intérieur de ces organites.
Déséquilibre énergétique et frénésie de construction d’ADN
L’accumulation d’ADP fait plus que signaler des centrales fatiguées. Elle alimente une voie chimique qui utilise des nutriments comme le glucose, la sérine et la glycine pour produire du formiate, un précurseur pour la synthèse de nucléotides puriques. Les chercheurs ont montré que lorsque le microARN-27b-5p ou HIF1α sont actifs, les cellules cardiaques intensifient cette voie de synthèse d’ADN et canalysent davantage de carbone issu de nutriments de base vers les acides nucléiques. Plutôt que d’encourager une division cellulaire normale, cet apport supplémentaire soutient des copies répétées de l’ADN sans séparation cellulaire, créant des cardiomyocytes à la fois plus volumineux et multinucléés. Des souris génétiquement modifiées pour surexprimer le microARN-27b dans leur muscle cardiaque ont développé des cœurs dilatés, des foyers de fibrose et une altération de la fonction de pompage, ressemblant de près à la pathologie humaine.
Bloquer la voie nuisible
Parce que le microARN se situe à un point de contrôle clé, les scientifiques ont testé si l’inhiber pouvait aider un cœur en échec à récupérer. Chez des souris soumises à une surcharge de pression sévère, ils ont utilisé des brins courts conçus pour neutraliser le microARN-27b-5p après l’apparition de l’insuffisance cardiaque. Ce traitement a restauré les niveaux d’ATP5A1, amélioré l’équilibre énergétique, réduit la proportion de cellules multinucléées, limité la fibrose et partiellement inversé l’augmentation cardiaque tout en améliorant la fonction de pompage. À la recherche d’une option médicamenteuse plus pratique, ils ont passé au crible des médicaments approuvés et identifié le méthotrexate, un antifolate déjà utilisé pour certains cancers et maladies auto-immunes, comme capable d’atténuer ce schéma de croissance délétère dans le cœur. Chez les souris stressées, le méthotrexate a réduit la copie nucléaire anormale et préservé la structure et la fonction cardiaques, vraisemblablement en restreignant la voie de synthèse d’ADN et en allégeant la contrainte en oxygène qui déclenche le microARN.
Ce que cela implique pour les patients
Cette étude montre qu’un signal de faible oxygène dans le cœur peut activer un microARN qui fragilise les centrales cellulaires, détourne des ressources vers la synthèse d’ADN et pousse les cellules cardiaques à croître de façon déformée et multinucléée, compromettant la fonction de pompe. En bloquant directement le microARN-27b-5p ou en utilisant des médicaments comme le méthotrexate pour limiter la voie de synthèse d’ADN hyperactive, il pourrait être possible de ralentir ou d’inverser cette hypertrophie nuisible. Bien que des travaux supplémentaires soient nécessaires avant de modifier les recommandations thérapeutiques, les résultats mettent en lumière une voie claire et pharmacologiquement accessible qui relie l’équilibre énergétique, l’apport en oxygène et la croissance du muscle cardiaque de manière compréhensible et potentiellement exploitable.
Citation: Mirtschink, P., Yuan, T., Bischof, C. et al. Hypoxia-driven microRNA-27b underlies pathologic cardiac endoreplication in heart disease. Sig Transduct Target Ther 11, 179 (2026). https://doi.org/10.1038/s41392-026-02656-x
Mots-clés: hypertrophie cardiaque, énergétique mitochondriale, microARN-27b, signalisation hypoxique, méthotrexate