Clear Sky Science · es
Base de datos y escalabilidad de aprendizaje profundo de propiedades de fonones anarmónicos mediante cálculos ab initio automatizados por fuerza bruta
Por qué importan las vibraciones que transportan calor
Cualquier sólido a nuestro alrededor, desde los chips de los teléfonos inteligentes hasta el aislamiento de los edificios, transporta calor principalmente mediante pequeñas vibraciones atómicas llamadas fonones. La facilidad con la que estas vibraciones se propagan determina si un material es adecuado para enfriar electrónica, mantener viviendas calientes o convertir calor residual en electricidad. Sin embargo, predecir con precisión este flujo térmico a partir de la estructura atómica de un material ha sido notoriamente difícil y lento. Este artículo presenta un nuevo enfoque para automatizar dichas predicciones para miles de cristales y luego emplear aprendizaje profundo para explorar espacios vastos de materiales posibles en busca de aquellos con conductividad térmica extremadamente alta o baja.
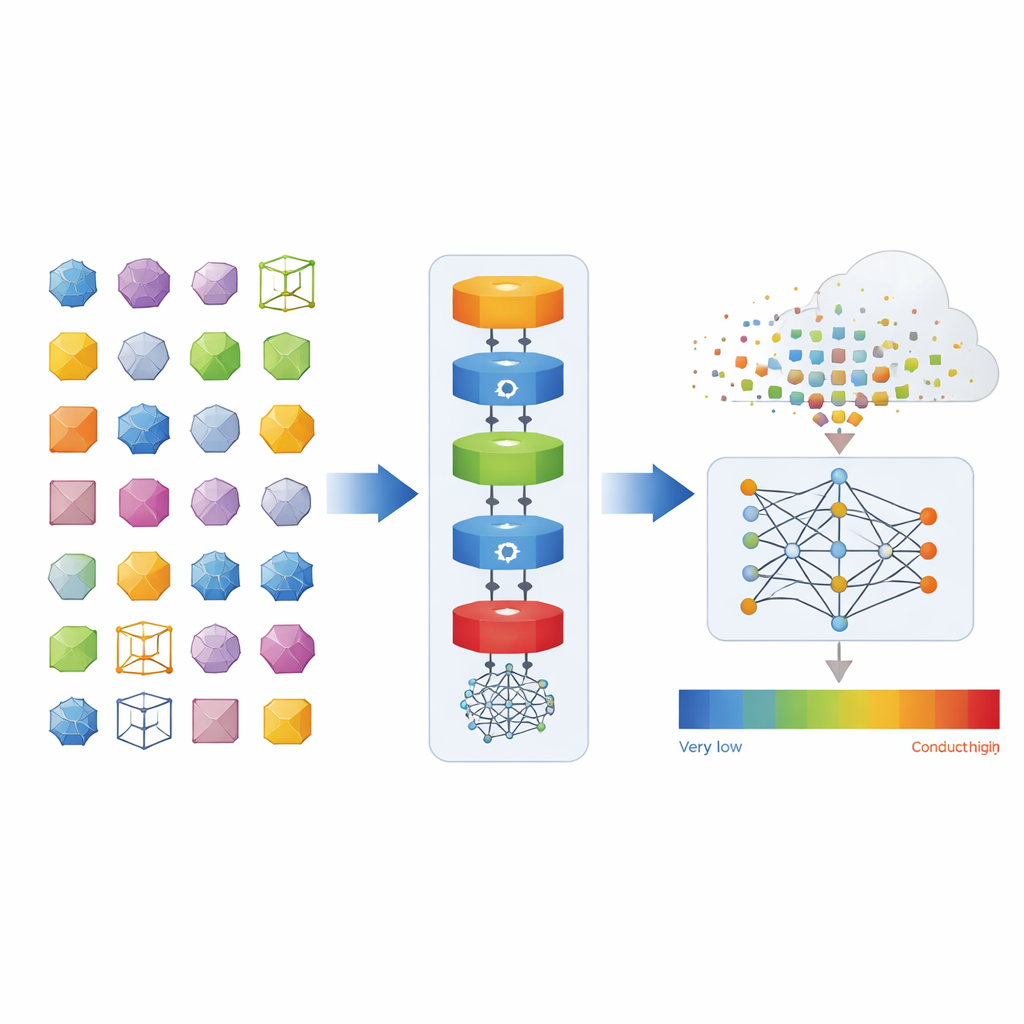
Construyendo una biblioteca gigante de cristales vibrantes
Los autores crearon un marco de software llamado auto-kappa que toma como entrada la estructura cristalina y ejecuta automáticamente una larga cadena de cálculos cuántico-mecánicos. Estos cálculos resuelven no solo cómo vibran los átomos de forma perfectamente elástica, sino también cómo las vibraciones colisionan y se dispersan de manera más realista y “desordenada”. A partir de esto, el software extrae propiedades detalladas, como las vidas medias de los fonones y cuánto contribuye cada vibración al transporte de calor. Usando esta canalización en superordenadores, el equipo ensambló una nueva base de datos, llamada Phonix, que cubre propiedades de fonones anarmónicos para más de 6500 cristales inorgánicos—desde sales simples como el cloruro de sodio hasta estructuras complejas con más de 100 átomos por celda unitaria.
Un mapa diverso de cómo los sólidos transportan calor
Con esta base de datos en mano, los investigadores examinaron cómo varía la conducción térmica entre distintos materiales. Encontraron que la conductividad térmica de la red, la fracción transportada por las vibraciones en la red atómica, tiende a disminuir a medida que crece el volumen por átomo—dicho de forma aproximada, las estructuras más abiertas suelen conducir peor el calor. La base de datos reveló una dispersión amplia: la mayoría de los materiales se sitúa entre aproximadamente 0,15 y 40 vatios por metro y grado kelvin a temperatura ambiente, pero una pequeña fracción alcanza valores extremadamente altos por encima de 200, y un subconjunto minúsculo supera 500 o incluso 1000. Muchos de los mejores conductores son formas de carbono y carburo de silicio, mientras que un gran número de compuestos exhibe conductividades térmicas bastante bajas, lo que ofrece ricas posibilidades para aplicaciones termoeléctricas o de aislamiento.
Contribuciones ondulatorias ocultas al flujo de calor
El calor en los cristales a menudo se imagina como fonones que se comportan como moléculas de gas, pero a escalas pequeñas las vibraciones también pueden actuar más como ondas superpuestas. La base de datos Phonix separa la contribución convencional “de tipo partícula” al transporte de calor de esta contribución más ondulatoria, o coherente. Para la mayoría de los materiales, especialmente los buenos conductores térmicos, predomina el canal tradicional de tipo partícula. Sorprendentemente, sin embargo, los autores encontraron muchos compuestos donde la parte coherente es considerable y en algunos casos comparable a la parte de partículas. Ciertas formas complejas de carburo de silicio, con muchos átomos y ramas de vibración densamente empaquetadas, muestran contribuciones coherentes particularmente grandes. Esto sugiere que el transporte de calor de tipo ondulatorio, a menudo ignorado en modelos prácticos, puede ser importante incluso en cristales con alta conductividad.
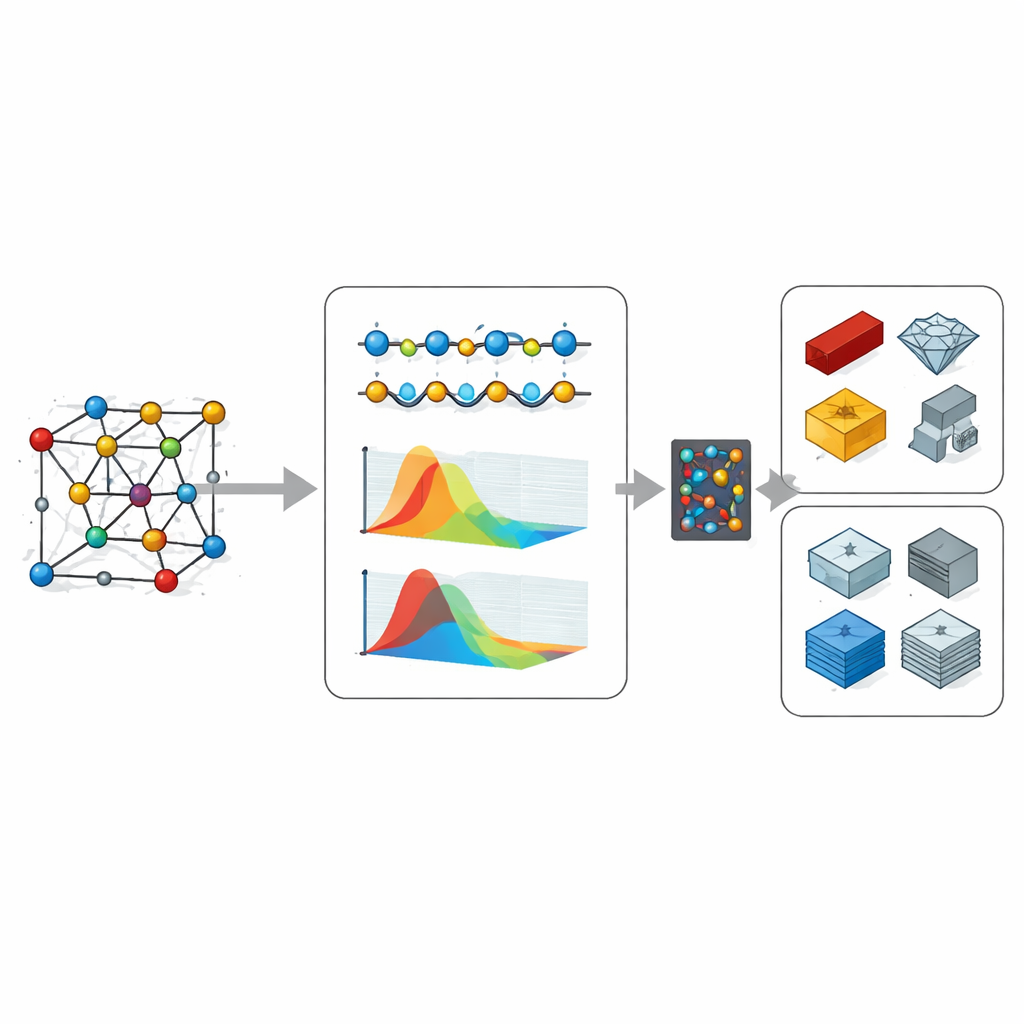
Enseñar a redes neuronales a leer planos atómicos
Para ir más allá de los costosos cálculos por fuerza bruta, el equipo entrenó redes neuronales basadas en grafos que toman la estructura atómica de un cristal y predicen su conductividad térmica—incluyendo cómo se acumula la contribución a medida que se incorporan vibraciones con diferentes longitudes de trayectoria media. Al variar el tamaño del conjunto de entrenamiento desde unos pocos cientos hasta varios miles de materiales, hallaron una ley de escalado clara: los errores de predicción disminuyen de forma predecible a medida que se añade más datos, similar a las tendencias observadas en modelos de lenguaje grande. Con estos modelos entrenados, los autores cribaron cientos de miles de cristales hipotéticos de la base de datos GNoME de DeepMind, y luego realizaron cálculos cuánticos completos en un subconjunto de los candidatos más prometedores para conductividades excepcionalmente altas o bajas.
Encontrar extremos: superconductores térmicos y superaislantes
El cribado descubrió nuevos materiales candidatos en los extremos de la conducción térmica. Algunos compuestos ricos en hidrógeno que contienen átomos muy pesados mostraron alta conductividad térmica porque sus vibraciones se separan claramente en modos de baja frecuencia de los átomos pesados y modos de alta frecuencia de los átomos ligeros, reduciendo la dispersión disruptiva. En el lado opuesto, estructuras complejas a base de cesio mostraron conductividades térmicas muy bajas, con vibraciones fuertemente mezcladas repartidas entre muchos átomos y frecuencias, lo que favorece la dispersión intensa y un pobre flujo de calor. Aunque algunos de estos cristales pueden ser difíciles de sintetizar, sus motivos estructurales compartidos ofrecen pistas valiosas para diseñar tanto “autopistas térmicas” altamente conductoras como “muros térmicos” altamente resistivos.
Qué significa esto para futuros materiales
En términos cotidianos, este trabajo aporta dos avances clave: una biblioteca amplia y de acceso abierto que captura cómo realmente se mueven y colisionan los átomos en miles de cristales, y un conjunto de modelos de aprendizaje automático que pueden leer esos planos atómicos para prever qué tan bien un material transportará calor. Juntos, ofrecen un atajo potente para descubrir mejores disipadores de calor para la electrónica, termoeléctricos mejorados para captura de energía y materiales avanzados para tecnologías en las que controlar el calor es tan importante como controlar la electricidad. A medida que la base de datos crezca e incluya efectos de vibración aún más sutiles, estas herramientas están en posición de hacer la búsqueda de nuevos materiales térmicos más rápida, más barata y mucho más sistemática que la experimentación por ensayo y error.
Cita: Ohnishi, M., Deng, T., Torres, P. et al. Database and deep-learning scalability of anharmonic phonon properties by automated brute-force first-principles calculations. npj Comput Mater 12, 150 (2026). https://doi.org/10.1038/s41524-026-02033-w
Palabras clave: conductividad térmica de la red, base de datos de fonones, vibraciones anarmónicas, informática de materiales, redes neuronales de grafos