Clear Sky Science · en
Ambiphilic behavior of hydrogen in trisubstituted silanes induced by substituent controlled polarity inversion
Why this tiny atom matters
Hydrogen atoms hide in almost every molecule around us, but they do not always behave the same way. In certain silicon-based compounds, a single hydrogen can sometimes act like an electron-rich "giver" and, in other cases, like an electron-poor "taker." This article explores why our usual ways of describing charge in molecules can miss this switch in personality and shows how a more complete map of electric forces around a molecule can reveal what hydrogen will actually do in a reaction.
Looking beyond simple charge labels
Chemists often assign each atom in a molecule a partial charge, a single number that says whether it is slightly positive or negative. These numbers are useful shortcuts, but they are averages over space and depend on the chosen model. Different charge schemes can give different answers for the same molecule and can blur out directional features that matter when two molecules approach each other. The authors argue that a better guide to reactivity is the molecular electrostatic potential, or ESP, which describes how a positive test charge would feel at each point in space around the molecule. This continuous landscape captures where the surroundings are electron-rich or electron-poor and in which directions those regions point.
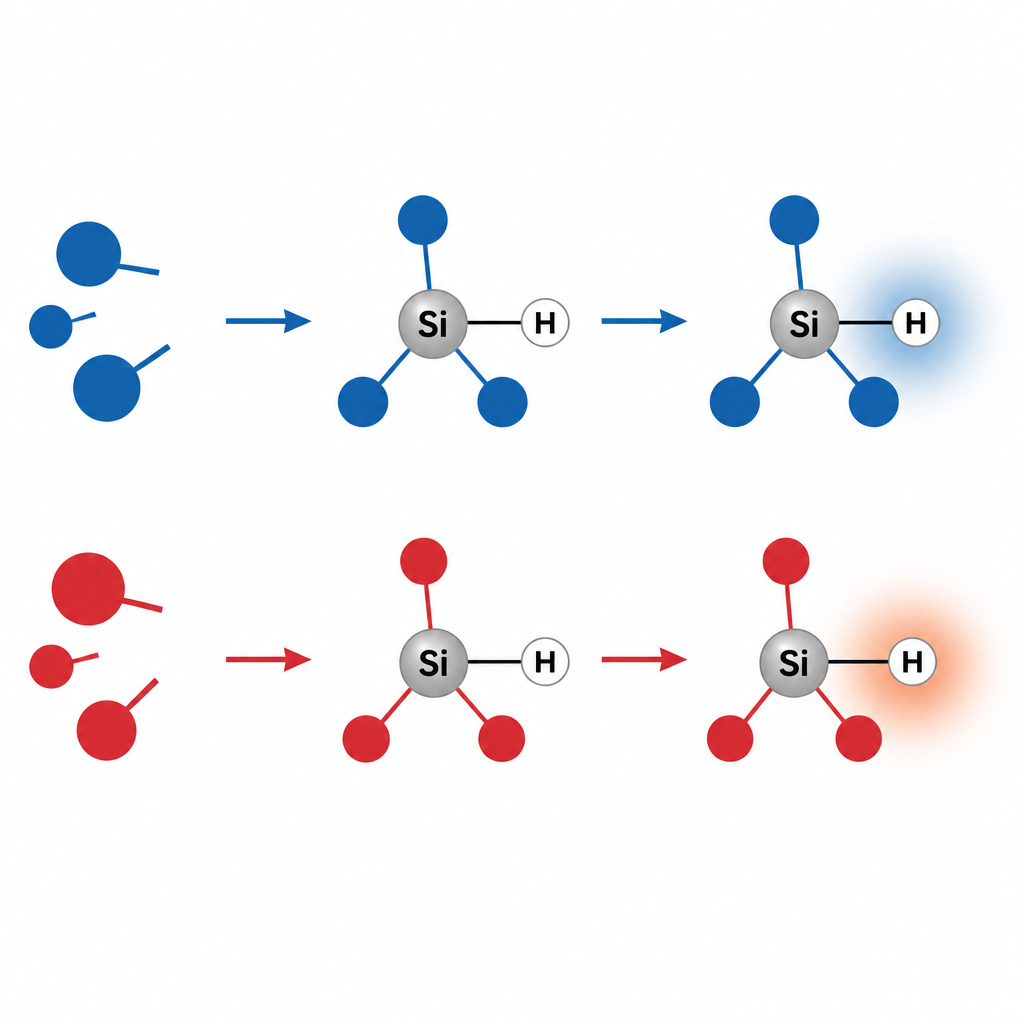
A surprising switch in silicon hydrides
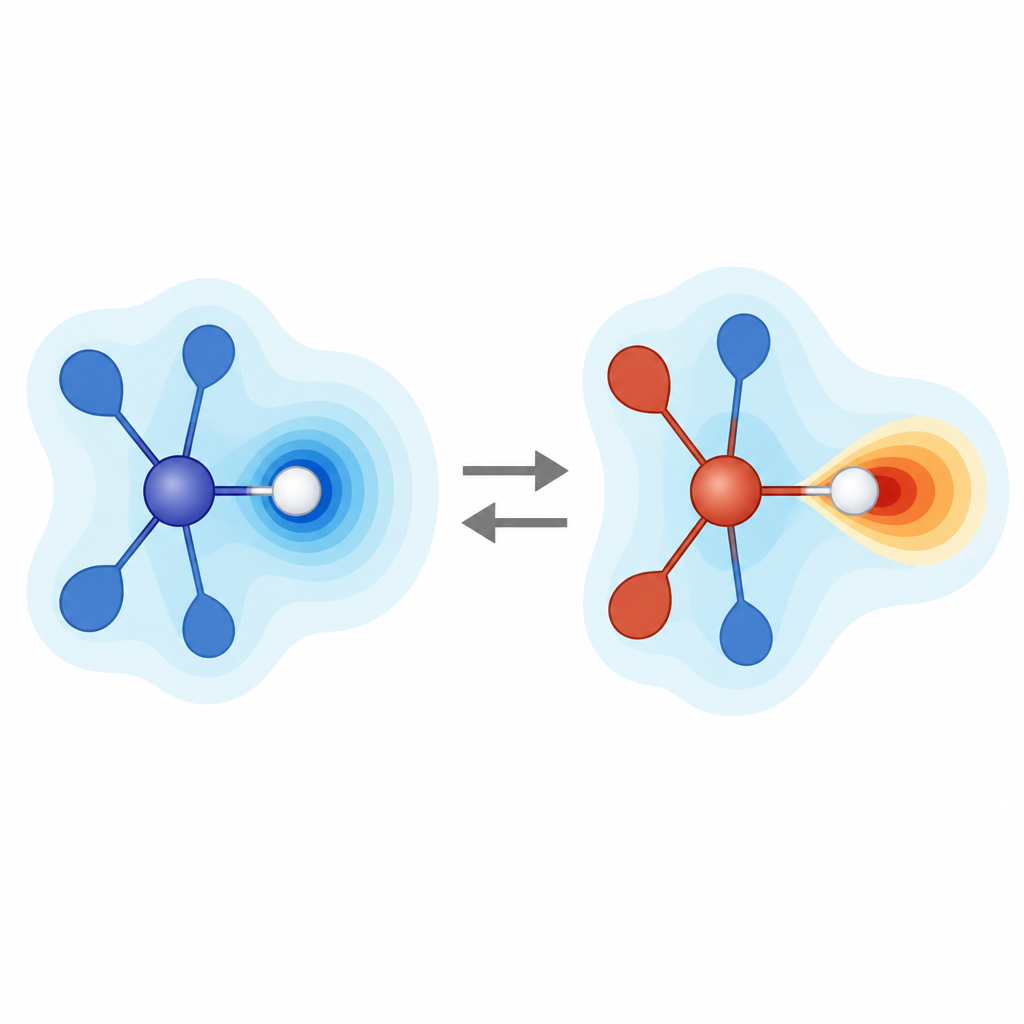
The study focuses on trisubstituted silanes, molecules where a silicon atom is bonded to three other groups and one hydrogen (Si–H). Simple electronegativity arguments say that silicon is less electronegative than hydrogen, so the bond should make hydrogen slightly negative, or "hydridic," in all cases. Indeed, all common charge models assign a negative charge to this hydrogen, even when the three attached groups pull strongly on electrons. But when the researchers examined the ESP at the surface near hydrogen, they found two very different patterns. With electron-donating groups, the region around hydrogen is negative, and hydrogen behaves as an electron-rich site. With electron-withdrawing groups, the local ESP near hydrogen turns positive, signaling an electron-poor, electrophilic character, even though the assigned atomic charge is still negative.
How the environment reshapes hydrogen
This inversion comes from the way the whole molecular framework redistributes electron density. Strongly pulling groups make the silicon center more positive and drain electron density along the Si–H bond axis, leaving a lobe of positive ESP pointing out from the hydrogen. This is reminiscent of the "sigma hole" seen on halogen atoms, where a positive region forms along a bond direction even on an overall negative atom. Here, however, hydrogen has no lone pairs to rearrange; the effect is collective and arises from the full molecule and its surroundings. The team confirmed that the same ESP-based picture also explains related bonds, such as aluminum–hydrogen, while more familiar carbon–hydrogen and phosphorus–hydrogen bonds remain consistently proton-like and electrophilic across the studied substitutions.
Testing the predictions in real liquids
To link the calculated electrostatic landscapes to measurable behavior, the authors measured proton nuclear magnetic resonance (NMR) signals of selected silanes in different solvents. As solvent polarity increased, silanes with electron-donating groups showed their Si–H signals shift upfield, indicating stronger electronic shielding and a more electron-rich hydrogen, in line with a more negative ESP. In contrast, silanes with electron-withdrawing groups shifted downfield in more polar solvents, consistent with an increasingly electron-poor hydrogen and more positive ESP. Detailed calculations across several solvents showed that changes in ESP, dipole moment, and NMR shift all track together, while simple atomic charges fail to mark the boundary between electron-rich and electron-poor behavior.
What this means for designing reactions
In everyday language, the work shows that the "weather map" of electric forces around a molecule is more revealing than a simple plus or minus sign stuck on each atom. In trisubstituted silanes, the shape of this map near hydrogen can be flipped by changing the three attached groups or the solvent, turning hydrogen from a donor of electron density into a seeker of it. This ambidextrous nature is rare for a neutral nonmetal system and has practical consequences for designing catalysts, predicting which partners a silane will bind to, and avoiding misleading conclusions drawn from partial charges alone. By using ESP as the main guide, chemists gain a clearer, more reliable way to tune and exploit the subtle behavior of silicon–hydrogen bonds.
Citation: Hrubý, V., Manna, D., Lo, R. et al. Ambiphilic behavior of hydrogen in trisubstituted silanes induced by substituent controlled polarity inversion. Commun Chem 9, 174 (2026). https://doi.org/10.1038/s42004-026-01980-1
Keywords: molecular electrostatic potential, silanes, hydrogen reactivity, sigma hole, NMR spectroscopy