Clear Sky Science · de
Ambiphiles Verhalten von Wasserstoff in trisubstituierten Silanen, ausgelöst durch substituenten-gesteuerte Polaritätsumkehr
Warum dieses winzige Atom wichtig ist
Wasserstoffatome stecken in fast jedem Molekül um uns herum, verhalten sich aber nicht immer gleich. In bestimmten siliziumbasierten Verbindungen kann ein einzelner Wasserstoff manchmal wie ein elektronreicher „Geber“ und in anderen Fällen wie ein elektronenarmer „Nehmer“ wirken. Dieser Artikel untersucht, warum unsere üblichen Beschreibungen elektrischer Ladung in Molekülen diese Persönlichkeitsänderung übersehen können, und zeigt, wie eine umfassendere Karte der elektrischen Kräfte um ein Molekül offenlegt, was der Wasserstoff tatsächlich in einer Reaktion tun wird.
Weiter denken als einfache Ladungszuschreibungen
Chemiker weisen oft jedem Atom in einem Molekül eine partielle Ladung zu, eine einzelne Zahl, die angibt, ob es leicht positiv oder negativ ist. Diese Zahlen sind nützliche Abkürzungen, aber sie sind Mittelwerte über den Raum und hängen vom gewählten Modell ab. Verschiedene Ladungsschemata können für dasselbe Molekül unterschiedliche Antworten geben und richtungsbezogene Merkmale verwischen, die wichtig sind, wenn sich zwei Moleküle annähern. Die Autoren argumentieren, dass ein besserer Wegweiser zur Reaktivität das molekulare elektrostatische Potential (ESP) ist, das beschreibt, wie eine positive Testladung an jedem Punkt im Raum um das Molekül herum empfinden würde. Diese kontinuierliche Landschaft erfasst, wo die Umgebung elektronereich oder elektronengering ist und in welche Richtungen diese Regionen weisen.
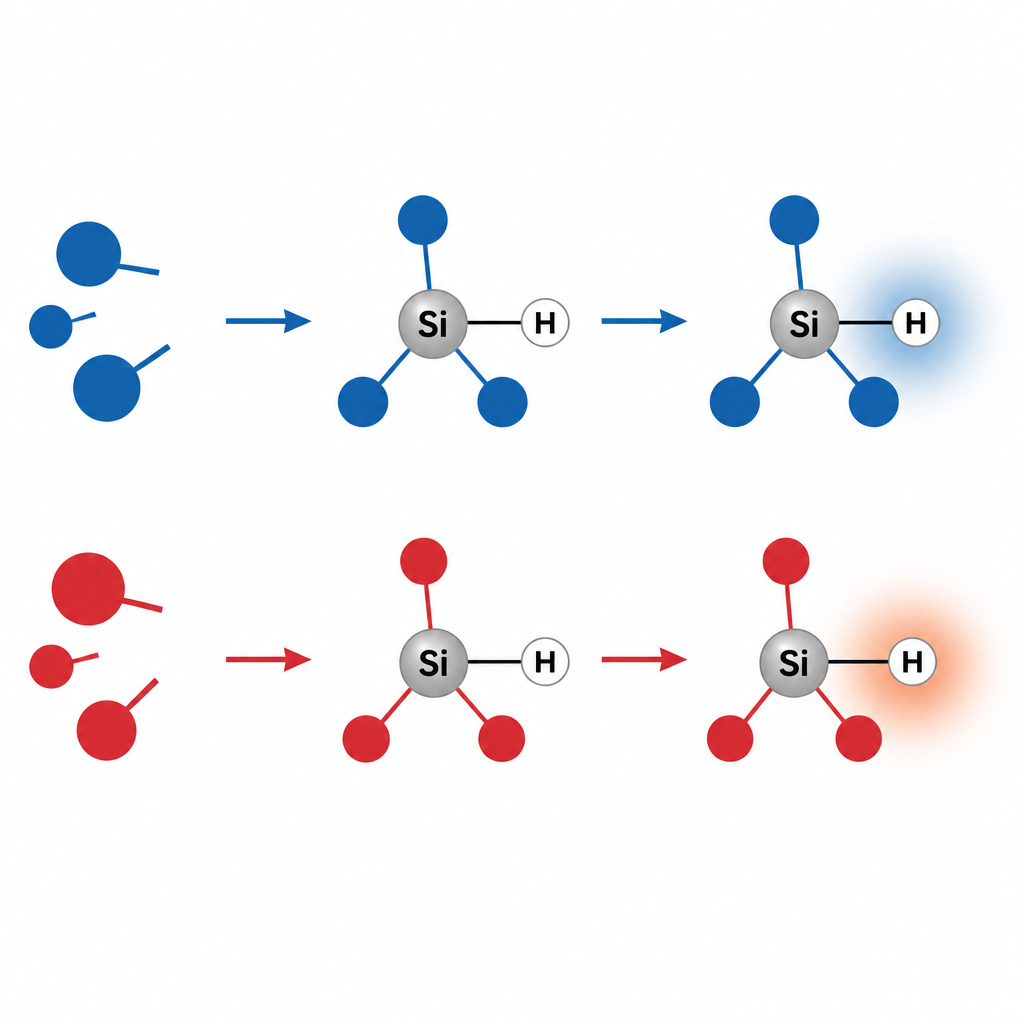
Ein überraschender Umschlag bei Silanhydriden
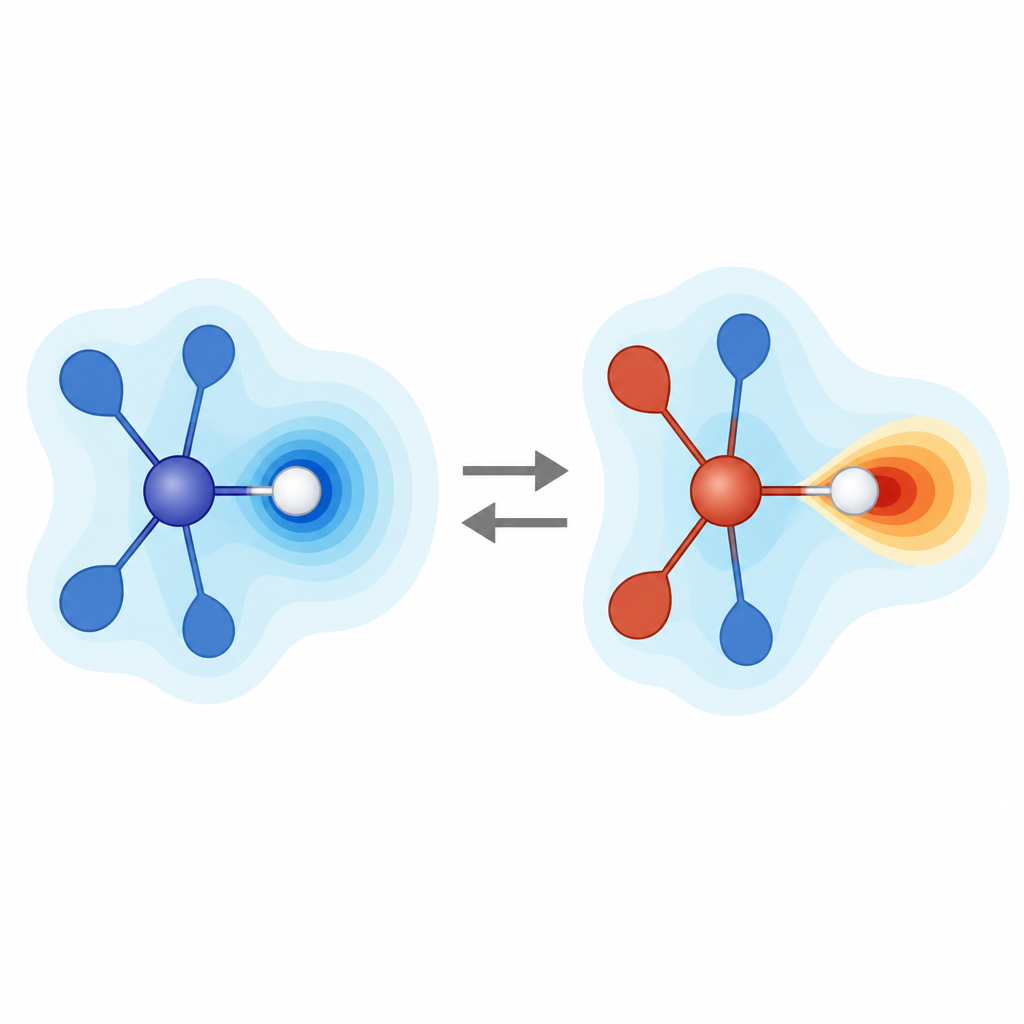
Die Studie konzentriert sich auf trisubstituierte Silane, Moleküle, bei denen ein Siliziumatom mit drei anderen Gruppen und einem Wasserstoff (Si–H) verbunden ist. Einfache Elektronegativitätsargumente besagen, dass Silizium weniger elektronegativ ist als Wasserstoff, sodass die Bindung den Wasserstoff in allen Fällen leicht negativ, also „hydridisch“, machen sollte. Tatsächlich weisen alle geläufigen Ladungsmodelle diesem Wasserstoff eine negative Ladung zu, selbst wenn die drei angehängten Gruppen stark an Elektronen ziehen. Als die Forscher jedoch das ESP an der Oberfläche in der Nähe des Wasserstoffs untersuchten, fanden sie zwei sehr unterschiedliche Muster. Bei elektronenspendeinduzierten Gruppen ist die Region um den Wasserstoff negativ, und der Wasserstoff verhält sich als elektronreicher Bereich. Bei elektronenziehenden Gruppen wird das lokale ESP in der Nähe des Wasserstoffs positiv, was eine elektronenarme, elektrophile Charakteristik anzeigt, obwohl die zugewiesene atomare Ladung weiterhin negativ ist.
Wie die Umgebung den Wasserstoff umformt
Diese Umkehr entsteht dadurch, wie das gesamte molekulare Gerüst die Elektronendichte umverteilt. Stark elektronenziehende Gruppen machen das Siliziumzentrum positiver und entziehen entlang der Si–H-Bindungsachse Elektronendichte, wodurch eine Wölbung positiven ESP vom Wasserstoff weg zeigt. Das erinnert an das „Sigma-Loch“, das bei Halogenatomen beobachtet wird, wo sich entlang der Bindungsrichtung eine positive Region bildet, selbst auf einem insgesamt negativen Atom. Hier hat Wasserstoff jedoch keine freien Elektronenpaare, die sich umordnen könnten; der Effekt ist kollektiv und entsteht aus dem gesamten Molekül und seiner Umgebung. Das Team bestätigte, dass dasselbe ESP-basierte Bild auch verwandte Bindungen wie Aluminium–Wasserstoff erklärt, während vertrautere Kohlenstoff–Wasserstoff- und Phosphor–Wasserstoff-Bindungen über die untersuchten Substitutionen hinweg durchweg protonenähnlich und elektrophil bleiben.
Vorhersagen in echten Flüssigkeiten testen
Um die berechneten elektrostatischen Landschaften mit messbarem Verhalten zu verknüpfen, maßen die Autoren Protonen-Kernspinresonanz(NMR)-Signale ausgewählter Silane in verschiedenen Lösungsmitteln. Mit zunehmender Polarität des Lösungsmittels zeigten Silane mit elektronenspendenden Gruppen eine Aufwärtverschiebung (upfield) ihrer Si–H-Signale, was stärkere elektronische Abschirmung und einen elektronereicheren Wasserstoff anzeigt—im Einklang mit einem negativeren ESP. Im Gegensatz dazu verschoben sich Silane mit elektronenziehenden Gruppen in polareren Lösungsmitteln nach unten (downfield), konsistent mit einem zunehmend elektronengeringen Wasserstoff und einem positiveren ESP. Detaillierte Rechnungen über mehrere Lösungsmittel zeigten, dass Änderungen im ESP, Dipolmoment und NMR-Verschiebung zusammenlaufen, während einfache atomare Ladungen die Grenze zwischen elektronereich und elektronengering nicht zuverlässig markieren.
Was das für das Design von Reaktionen bedeutet
Einfach gesagt zeigt die Arbeit, dass die „Wetterkarte“ der elektrischen Kräfte um ein Molekül aufschlussreicher ist als ein einfaches Plus- oder Minuszeichen an jedem Atom. Bei trisubstituierten Silanen kann die Form dieser Karte in der Nähe des Wasserstoffs durch Änderung der drei angehängten Gruppen oder des Lösungsmittels umgekehrt werden, sodass der Wasserstoff vom Geber von Elektronendichte zum Sucher danach wird. Diese ambivalente Natur ist für ein neutrales Nichtmetallsystem selten und hat praktische Konsequenzen für das Design von Katalysatoren, die Vorhersage, mit welchen Partnern ein Silan bindet, und das Vermeiden irreführender Schlussfolgerungen, die allein aus partiellen Ladungen gezogen werden. Indem Chemiker das ESP als Hauptleitfaden nutzen, erhalten sie einen klareren, verlässlicheren Weg, das subtile Verhalten von Si–H-Bindungen zu steuern und zu nutzen.
Zitation: Hrubý, V., Manna, D., Lo, R. et al. Ambiphilic behavior of hydrogen in trisubstituted silanes induced by substituent controlled polarity inversion. Commun Chem 9, 174 (2026). https://doi.org/10.1038/s42004-026-01980-1
Schlüsselwörter: molekulares elektrostatisches Potential, Silikane, Wasserstoffreaktivität, Sigma-Loch, NMR-Spektroskopie