Clear Sky Science · en
Shared patterns of dysregulated gene expression across squamous cell carcinomas unveil predictors for prognosis and drug sensitivity
Why this matters for cancer patients
Cancers that start from the body’s lining cells—called squamous cells—can appear in the skin, mouth and throat, esophagus, lungs, and cervix. Although they arise in very different organs, they kill hundreds of thousands of people each year. This study asks a simple but powerful question: under the surface, do these squamous cell cancers behave so similarly that we can predict which patients will do poorly and which drugs might work best, just by reading patterns in their gene activity?
Looking for common threads across many cancers
The researchers gathered gene activity data from 1,790 samples of tumors and nearby noncancerous tissue from five major squamous cell cancers: those of the lung, head and neck, esophagus, cervix, and skin. Using both older microarray technology and newer RNA sequencing, they measured how strongly thousands of genes were turned on or off in tumors compared with their neighboring normal tissue. They then asked how similar these “dysregulated” patterns were across cancer types, and compared them with two related but distinct cancers, lung and esophageal adenocarcinomas, which arise from different cell types.
They found that the five squamous cancers share strongly overlapping patterns of gene disruption, far more so than the overlaps between squamous cancers and adenocarcinomas in the same organ. In particular, head and neck, esophageal, and cervical squamous cancers looked strikingly alike at the gene-expression level, while lung and skin squamous cancers showed more distinct molecular personalities. These results held up when tested in completely independent datasets, suggesting that there is a robust “squamous program” of gene activity that cuts across organ boundaries.
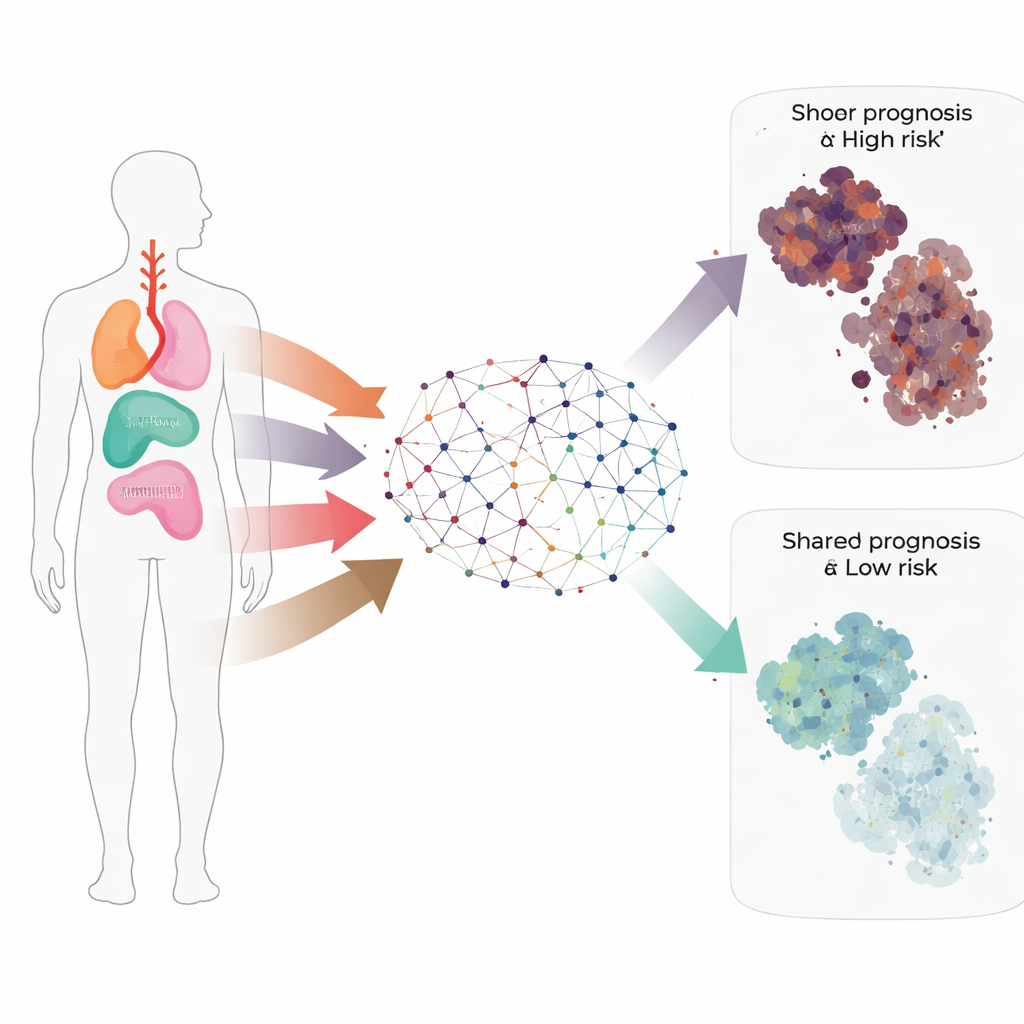
Gene networks and hidden subtypes
To move beyond lists of genes, the team built a co-expression network—a kind of map showing which genes rise and fall together across hundreds of samples. This analysis grouped genes into nine modules, each tied to particular biological jobs such as copying DNA, burning fuel, remodeling the tissue scaffold, forming blood vessels, or coordinating immune responses. From these modules, the researchers pulled out 441 “hub” genes that sit at the centers of these networks and are likely to influence many other genes. Many are already known troublemakers in squamous cancers, such as SOX2, TP63, and COL1A1.
Using the activity of these hub genes, they then reclassified head and neck, esophageal, and cervical squamous cancers into four molecular subtypes. These subtypes cut across the original organ labels and showed clear differences in patient age, sex, infection with human papillomavirus, and, most importantly, survival. One subtype in particular (Subtype 3) stood out for its much better prognosis, while the other three subtypes had consistently worse outcomes.
Balancing invasion and the immune system
The puzzle was why some subtypes fare so much worse. By comparing the gene activity between the good-outcome subtype and the poor-outcome subtypes, the study highlighted two opposing forces. The aggressive subtypes showed strong signals of “epithelial–mesenchymal transition” (EMT) and remodeling of the extracellular matrix—the processes that help cancer cells loosen from their neighbors and invade surrounding tissue. In contrast, the favorable subtype was enriched for genes linked to immune activity, including T cells and other defenders that can recognize and attack tumors.
Diving deeper, the team focused on key EMT-driving genes such as SNAI2 and TWIST1. Higher levels of these genes went hand-in-hand with lower levels of anti-tumor immune cells like CD8 and CD4 T cells and activated dendritic cells inside the tumors. Patients whose tumors had more SNAI2 or TWIST1 tended to live for a shorter time. These findings support a picture in which invasive, shape-shifting cancer cells help create an immunosuppressive neighborhood, blunting the body’s natural defenses and worsening prognosis.
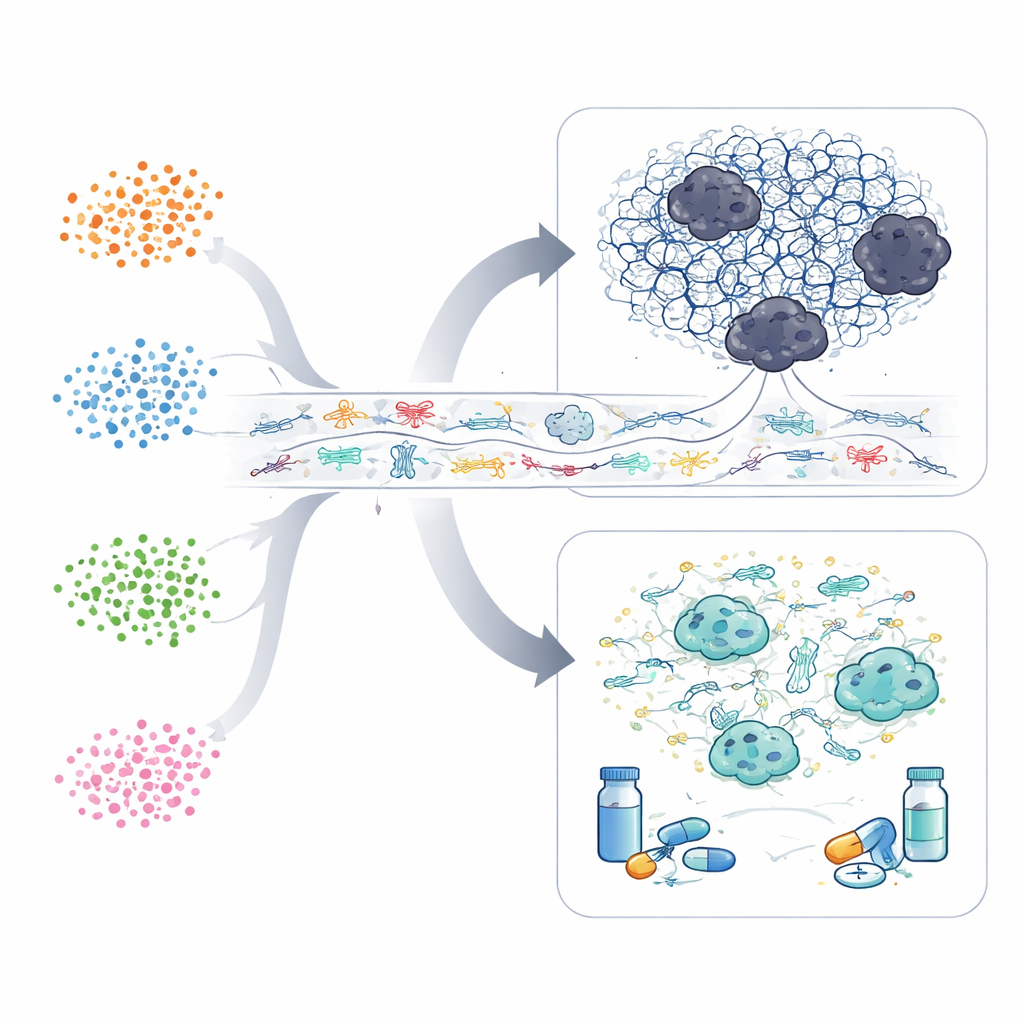
A six-gene guide to risk and treatment
Finally, the researchers searched for a small set of genes that could reliably flag high-risk patients. They zeroed in on six genes—COL1A1, MMP1, SERPINE1, KRT6A, IGF2BP3, and SPP1—that were more active in the worst-outcome subtype and whose high expression was linked to shorter survival. Using only these six measurements, they built a risk score that separated patients into low- and high-risk groups. Across multiple datasets, those in the high-risk quarter of scores died much sooner than those in the low-risk quarter.
When the team linked this risk score to predicted responses to common chemotherapy drugs, they found distinct patterns: low-risk patients appeared more sensitive to drugs such as cisplatin, afatinib, gemcitabine, and irinotecan, while high-risk patients seemed more responsive to vinblastine and vinorelbine. This suggests that the same six-gene signature that signals danger might also hint at which medicines are most likely to help.
What this means for patients
For a non-specialist, the key message is that many squamous cell cancers—regardless of where they arise—share a deep molecular script. By reading this script in the form of gene activity patterns, scientists can sort patients into groups with very different risks and likely drug responses. The six-gene signature uncovered here is not yet a clinical test, and the findings still need lab and prospective clinical validation. But the work shows how large-scale data can uncover common weak points in diverse cancers and points toward more personalized, gene-guided treatment strategies for people facing squamous cell carcinoma.
Citation: Wang, D., Li, X., Zhou, J. et al. Shared patterns of dysregulated gene expression across squamous cell carcinomas unveil predictors for prognosis and drug sensitivity. Sci Rep 16, 12833 (2026). https://doi.org/10.1038/s41598-026-41052-x
Keywords: squamous cell carcinoma, gene expression, cancer subtypes, tumor immune microenvironment, prognostic biomarkers