Clear Sky Science · en
Simulating electron transfer on noisy quantum computers
Why this matters for future energy tech
Many next‑generation batteries, solar cells and quantum devices rely on tiny, ultra‑fast motions of electrons and atoms that are extremely hard to calculate on ordinary computers. This paper shows how today’s noisy quantum computers can already mimic a key piece of that story: how an electron moves through a molecular network while jostling against local vibrations, the kind of motion that ultimately controls efficiency and heat loss in real materials.
Electrons, vibrations and the problem of heat
In energy materials, such as organic solar cells or battery electrodes, electrons rarely travel alone. As they hop between molecular sites, they tug on nearby atoms, creating vibrations that store and release energy. These coupled motions can keep electronic states "in step" with specific vibrations for surprisingly long times, helping charges separate quickly instead of getting stuck and wasted as heat. Standard equilibrium theories often fail in these situations, especially when the environment damps vibrations only slowly. Capturing these non‑equilibrium effects is crucial for designing devices that move charge efficiently while minimizing energy loss.
Turning hardware noise into a useful feature
The authors build on a mathematical trick called the pseudomode approach, which replaces a complex vibrational environment with a small set of damped oscillators. Each electronic site in their model gets its own local oscillator that captures how a specific high‑frequency molecular vibration shapes charge motion. On the quantum processor, each site is encoded in one qubit and each oscillator in another, giving a simple, regular layout. A key insight is that the natural decay of qubits, usually treated as a nuisance, can stand in for the damping of these oscillators. By choosing qubits with suitable lifetimes and carefully slicing time into small steps, the team lets the hardware’s own dissipation emulate vibrational relaxation, while a custom error‑filtering scheme removes other noise that does not match the physical model. 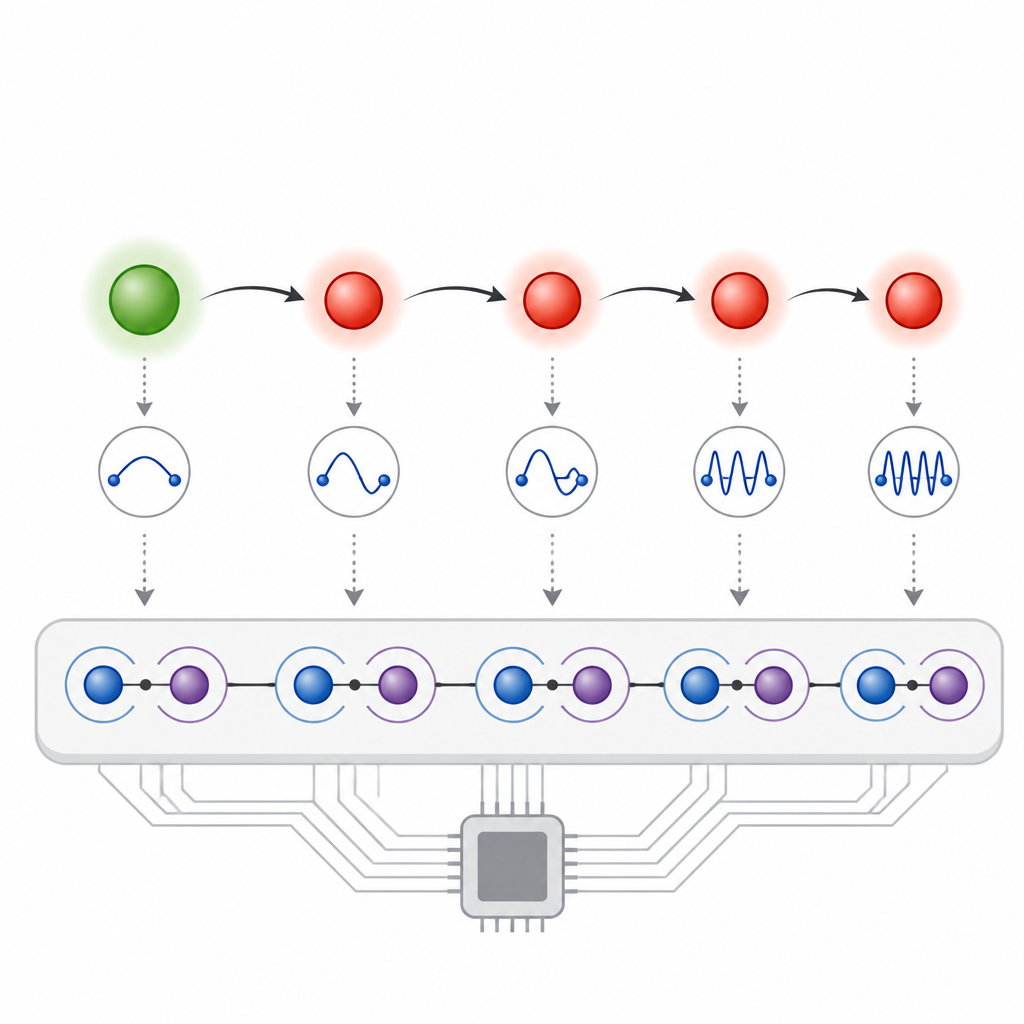
Testing fast transfer routes in a model chain
To test this strategy, the researchers model a chain of molecular sites with one donor at one end, a nearby energy trap and a string of acceptors. An electron starts on the donor and can either fall into the trap or escape along the chain. By tuning the energy offset between donor and acceptors, they probe two routes. In the purely electronic case, transfer occurs when the donor energy aligns directly with an acceptor state. In the vibronic case, the donor instead lines up with an acceptor state plus one quantum of vibration, so the electron and the local vibration act together. On an IBM superconducting device, they simulate these dynamics with up to 10 electronic sites and 10 oscillators, and compare the measured site populations over time with high‑precision classical calculations. Distinct peaks in the time‑averaged transfer probability reveal both electronic and vibronic resonance conditions, and the quantum hardware clearly distinguishes runs with and without electron‑vibration coupling.
Following entangled motion at larger scales
The work goes beyond spotting peaks. By examining how the electron population builds up on different sites, the authors show that vibronic coupling supports a ratchet‑like drift of charge away from the trap, creating a long‑lived, non‑equilibrium state that favors separation over recombination. They then scale the model from 3 to 10 sites, keeping the circuit depth almost constant by arranging gates in parallel layers. For each size, they run many experiments and benchmark against noiseless simulations. A tailored post‑selection step discards measurement shots that break simple conservation rules, stripping away depolarizing errors while leaving the intended damping intact. Across sizes, the best runs match a model with vibrational lifetimes of roughly 50–150 femtoseconds, approaching the lifetimes of key high‑frequency bond‑stretching modes in real organic molecules. 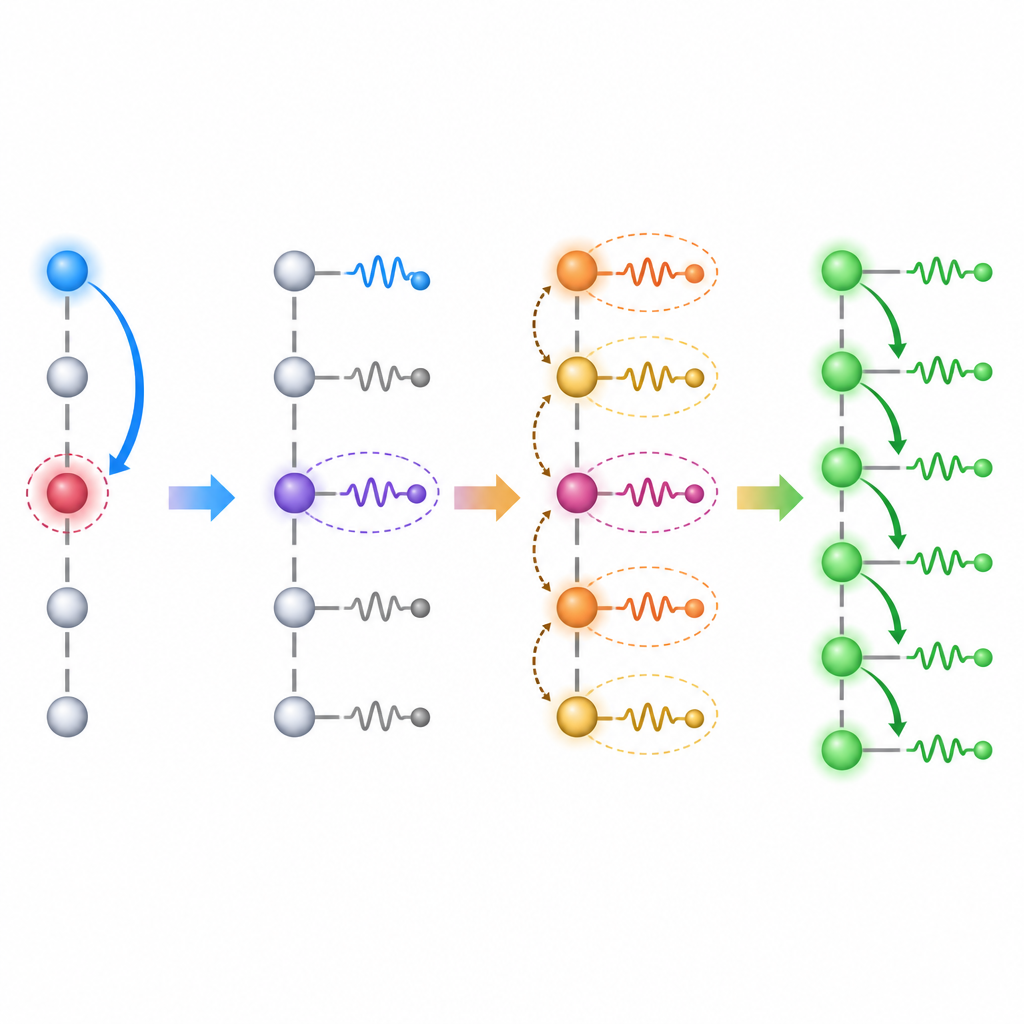
What this means for quantum simulations
The study demonstrates that present‑day noisy quantum processors can already reproduce subtle charge‑transfer signatures that depend on sustained entanglement between electronic sites and local vibrations. Rather than demanding perfectly isolated qubits, the method embraces certain kinds of hardware loss as part of the physics to be simulated, while filtering out incompatible noise through the structure of the model itself. Because the required circuit depth does not grow with system size, the approach offers a practical path toward simulating larger vibronic networks and more complex environments. In plain terms, the authors turn some of the messiness of real quantum hardware into a tool, showing that even imperfect quantum computers can help us understand how electrons and vibrations cooperate to move energy through advanced materials.
Citation: Gajewski, M., Somoza, A.D., Schmiedinghoff, G. et al. Simulating electron transfer on noisy quantum computers. Nat Commun 17, 4779 (2026). https://doi.org/10.1038/s41467-026-73700-1
Keywords: electron transfer, vibronic coupling, noisy quantum computers, open quantum systems, energy materials