Clear Sky Science · en
Integrating ANI and phylogenies for re-evaluation of Fusobacterium taxonomy and disease associations
Why these mouth and gut microbes matter
Microbes from the genus Fusobacterium live in our mouths and intestines, and have recently been linked to diseases ranging from gum infections to colorectal cancer. Yet even experts have struggled to agree on where one Fusobacterium species ends and another begins, and different naming systems have muddled the picture. This study tackles that confusion head‑on by using modern genome analysis to redraw the family tree of Fusobacterium and to build simple tools any lab can use to tell closely related species apart—work that directly affects how we understand, diagnose and eventually treat diseases tied to these bacteria. 
Untangling a confusing family of microbes
For years, many disease studies have focused on a catch‑all group called Fusobacterium nucleatum. Historically it was divided into subspecies with names such as animalis, polymorphum, vincentii and nucleatum, but genetic evidence hinted that these subspecies were different enough to be full species. On top of that, new clades and labels—like Fna C1, Fna C2, and F. watanabei—were introduced by different research groups using varying methods. As a result, the same lineage might be called by several names in the literature, making it hard to compare studies or pin specific disease risks on specific bacterial types.
Reading the genomes to draw clear boundaries
The authors assembled 540 high‑quality Fusobacterium genomes from public databases and compared them using a measure called average nucleotide identity (ANI), which captures how similar two genomes are overall. When they plotted all pairwise comparisons, a striking gap appeared in the data: no genome pairs had ANI values in a narrow band around roughly 93.4–93.9%. Above this gap, genomes grouped cleanly into clusters; below it, they formed clearly separate ones. A whole‑genome phylogenetic tree—essentially a family tree built from many genetic differences—lined up closely with these ANI clusters. Together, these results show that the ANI gap acts as a natural dividing line between Fusobacterium species, allowing the authors to sort all 540 genomes into 34 well‑defined species, including six newly proposed ones.
Fixing names and discovering new neighbors
Armed with this genus‑wide view, the team re‑examined how genomes were labeled in reference databases. Nearly one in five strains had names that needed correction or updating. Crucially, the work confirms that the old “F. nucleatum” subspecies vincentii, polymorphum, nucleatum (sensu stricto), and the animalis clades C1 and C2 are in fact distinct species—including F. animalis and F. watanabei. The analysis also revealed new species, such as Fusobacterium sp. bovis from cattle lesions and F. heteroulcerans, which had previously been lumped under ulcerans or varium. In some cases, the authors showed that certain lineages sit so far from other Fusobacterium that they may eventually warrant their own genera, highlighting unexpectedly deep evolutionary splits inside this group.
Simple genetic shortcuts for precise identification
Whole‑genome sequencing is powerful but too costly and complex for routine use in many clinical labs. Traditionally, researchers have relied on the 16S rRNA gene as a barcode, but within Fusobacterium this gene is too conserved—and often present in multiple slightly different copies—to reliably distinguish species. The authors instead tested three single‑copy genes, gyrB, rpoB and znpA, and found that gyrB and rpoB in particular track the whole‑genome relationships very closely. They then defined short variable segments within gyrB and rpoB that can be amplified by standard PCR and compared to a curated reference set. Using a stepwise “B&B” strategy—first gyrB, then rpoB if needed—they were able to assign species for 45 clinically relevant strains with perfect agreement to full‑genome ANI results, including colorectal cancer and oral isolates.
Re‑reading cancer microbiome studies with better labels
To show why correct naming matters, the team plugged their revised taxonomy into popular genomic and metagenomic tools, including the Genome Taxonomy Database and MetaPhlAn, which many groups use to profile microbiomes from stool or tissue samples. By remapping species‑level genome bins (SGBs) used in large colorectal cancer studies, they discovered that several bacterial signals originally reported broadly as “F. nucleatum” actually correspond to distinct species such as F. animalis, F. vincentii, F. polymorphum, F. nucleatum sensu stricto and F. watanabei. This finer resolution lets researchers ask which exact species are enriched in tumors, which colonize healthy tissue, and how their roles differ in driving inflammation, immune responses or treatment resistance. 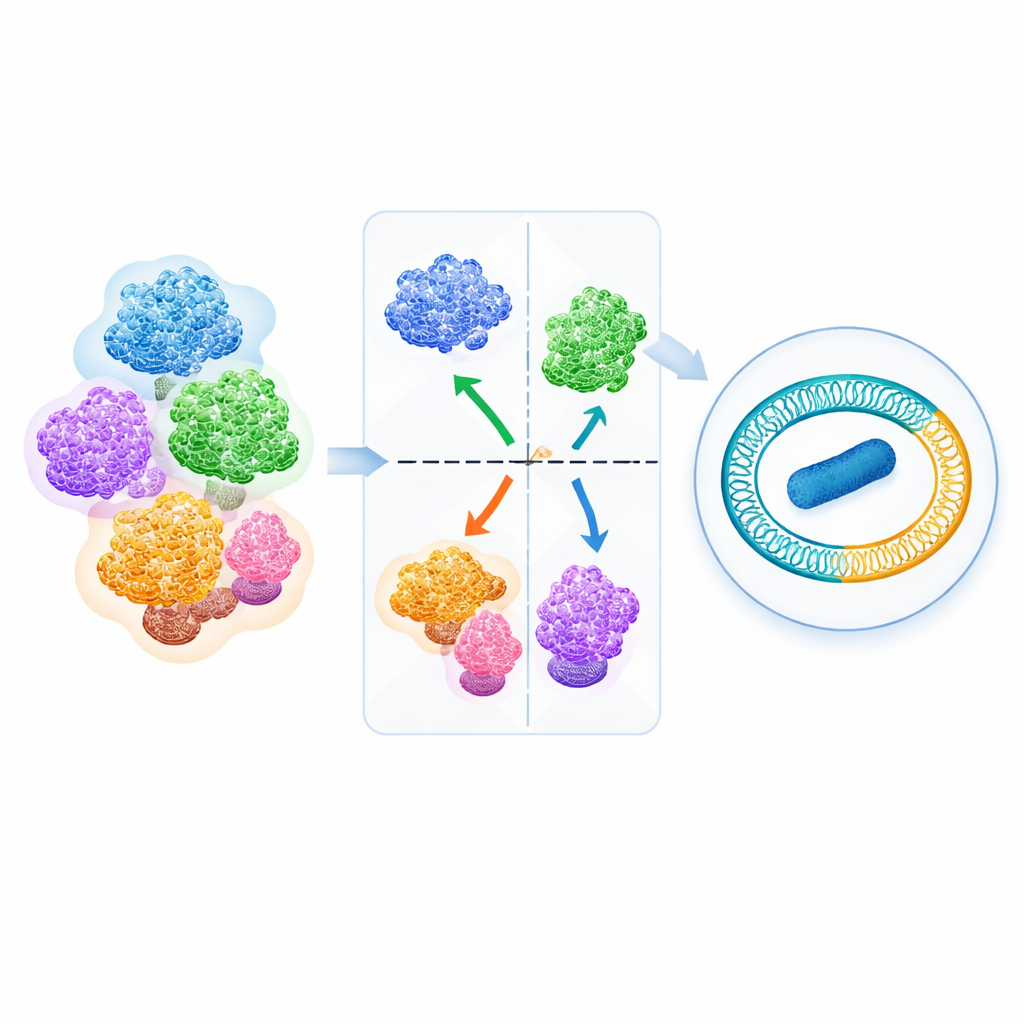
What this means for future research and care
In practical terms, this work replaces a fuzzy picture of “F. nucleatum” with a sharp map of 34 genetically distinct Fusobacterium species and offers a simple two‑gene test to tell them apart without sequencing entire genomes. For clinicians and researchers, that means clearer links between specific microbes and specific disease outcomes, more reliable comparisons across studies, and better targeting of diagnostics or therapies aimed at Fusobacterium. As more genomes are added, the exact boundary may be refined, but the framework established here—using a natural genomic gap plus robust marker genes—provides the taxonomic precision needed to advance both basic and clinical research on how these microbes influence infections, cancer and beyond.
Citation: Bi, D., Wu, Y., Ji, G. et al. Integrating ANI and phylogenies for re-evaluation of Fusobacterium taxonomy and disease associations. Nat Commun 17, 3774 (2026). https://doi.org/10.1038/s41467-026-70540-x
Keywords: Fusobacterium taxonomy, microbiome and cancer, bacterial species identification, genome-based classification, colorectal cancer microbiota