Clear Sky Science · de
Integration von ANI und Phylogenien zur Neubewertung der Fusobacterium-Taxonomie und krankheitsbezogener Assoziationen
Warum diese Mund‑ und Darmmikroben wichtig sind
Microben der Gattung Fusobacterium leben in Mund und Darm und werden mittlerweile mit Erkrankungen von Zahnfleischinfektionen bis hin zu kolorektalem Krebs in Verbindung gebracht. Selbst Fachleute hatten jedoch Schwierigkeiten, zu vereinbaren, wo eine Fusobacterium‑Art endet und eine andere beginnt; verschiedene Benennungssysteme haben das Bild zusätzlich verwischt. Diese Studie geht das Problem direkt an, indem sie moderne Genomanalysen nutzt, um den Stammbaum von Fusobacterium neu zu zeichnen, und einfache Werkzeuge entwickelt, die jedes Labor verwenden kann, um eng verwandte Arten zu unterscheiden — Arbeit, die direkt beeinflusst, wie wir Erkrankungen, die mit diesen Bakterien zusammenhängen, verstehen, diagnostizieren und schließlich behandeln.
Eine verworrene Mikrobenfamilie entwirren
Jahrelang konzentrierten sich viele Krankheitsstudien auf eine Sammelgruppe namens Fusobacterium nucleatum. Historisch war sie in Subspecies wie animalis, polymorphum, vincentii und nucleatum unterteilt, doch genetische Hinweise deuteten darauf hin, dass diese Subspecies genug Unterschiede aufweisen, um als eigene Arten zu gelten. Darüber hinaus führten verschiedene Forschungsgruppen mit unterschiedlichen Methoden neue Kladen und Bezeichnungen ein — etwa Fna C1, Fna C2 und F. watanabei. Infolgedessen kann dieselbe Abstammungslinie in der Literatur unter mehreren Namen auftreten, was den Vergleich von Studien oder die Zuordnung spezifischer Krankheitsrisiken zu bestimmten bakteriellen Typen erschwert.
Die Genome lesen, um klare Grenzen zu ziehen
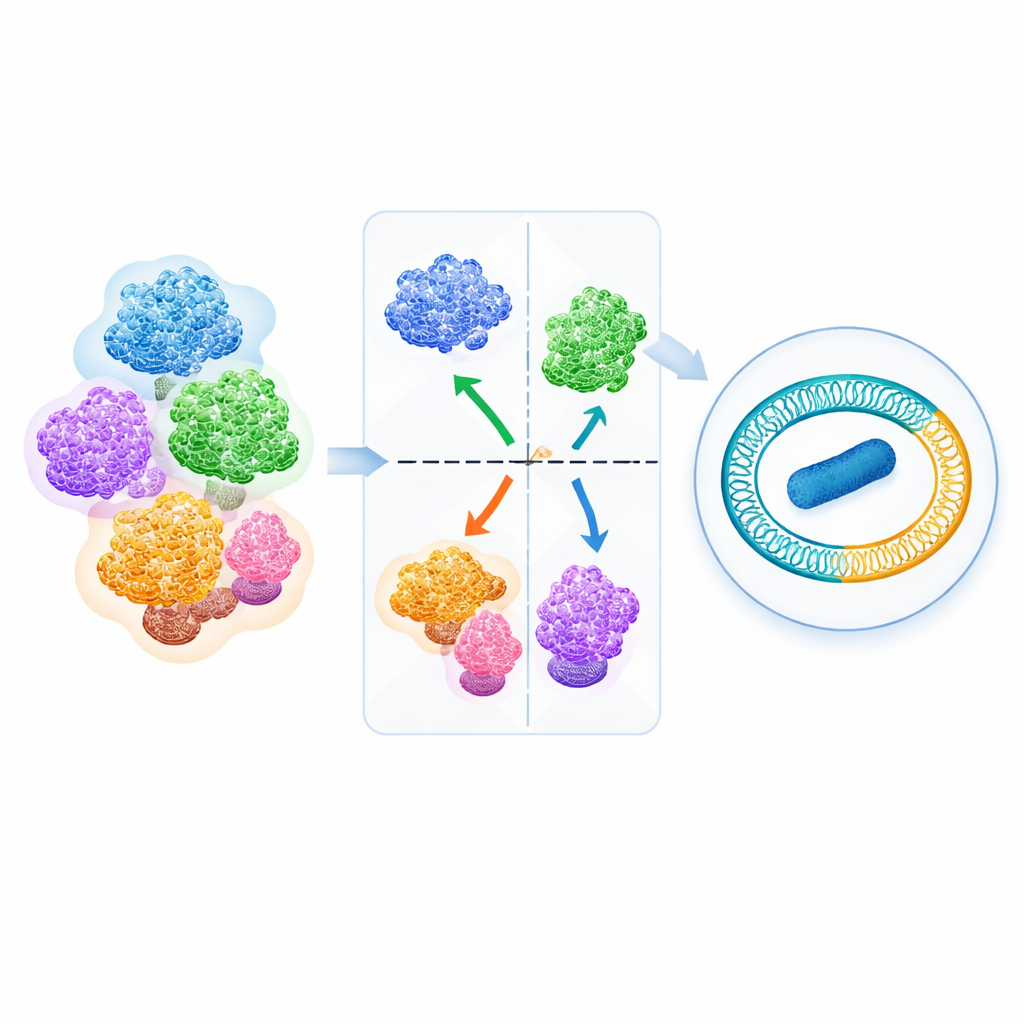
Die Autor*innen versammelten 540 hochwertige Fusobacterium-Genome aus öffentlichen Datenbanken und verglichen sie mit einem Maß namens Average Nucleotide Identity (ANI), das die Gesamtähnlichkeit zweier Genome erfasst. Beim Plotten aller paarweisen Vergleiche zeigte sich eine markante Lücke in den Daten: Es gab keine Genompaarwerte in einem schmalen Bereich von etwa 93,4–93,9% ANI. Oberhalb dieser Lücke gruppierten sich Genome sauber zu Clustern; darunter bildeten sie klar getrennte Gruppen. Ein Ganzgenom-phylogenetischer Baum — im Grunde ein Stammbaum, der aus vielen genetischen Unterschieden konstruiert ist — stimmte eng mit diesen ANI-Clustern überein. Zusammen zeigen diese Ergebnisse, dass die ANI-Lücke als natürliche Trennlinie zwischen Fusobacterium‑Arten fungiert und es den Autor*innen ermöglichte, alle 540 Genome in 34 gut definierte Arten einzuteilen, darunter sechs neu vorgeschlagene.
Namen korrigieren und neue Verwandte entdecken
Mit dieser gattungweiten Sicht überprüfte das Team die Benennungen in Referenzdatenbanken. Nahezu einer von fünf Stämmen trug einen Namen, der berichtigt oder aktualisiert werden musste. Entscheidend bestätigt die Arbeit, dass die früheren „F. nucleatum“-Subspecies vincentii, polymorphum, nucleatum (sensu stricto) und die animalis‑Kladen C1 und C2 tatsächlich eigenständige Arten sind — einschließlich F. animalis und F. watanabei. Die Analyse deckte außerdem neue Arten auf, etwa Fusobacterium sp. bovis aus Rinderläsionen und F. heteroulcerans, die zuvor unter ulcerans oder varium zusammengefasst worden waren. In einigen Fällen zeigten die Autor*innen, dass bestimmte Linien so weit von anderen Fusobacterium entfernt liegen, dass sie möglicherweise schließlich eigene Gattungen rechtfertigen — ein Hinweis auf unerwartet tiefe evolutionäre Aufspaltungen innerhalb dieser Gruppe.
Einfache genetische Abkürzungen für präzise Identifikation
Ganzgenomsequenzierung ist leistungsfähig, aber für den Routineeinsatz in vielen klinischen Laboren zu teuer und zu komplex. Traditionell nutzten Forschende das 16S-rRNA‑Gen als Barcode, doch innerhalb von Fusobacterium ist dieses Gen zu konserviert — und oft in mehreren leicht unterschiedlichen Kopien vorhanden —, um Arten zuverlässig zu unterscheiden. Die Autor*innen testeten stattdessen drei Single‑Copy-Gene, gyrB, rpoB und znpA, und fanden, dass insbesondere gyrB und rpoB die Ganzgenom‑Beziehungen sehr gut abbilden. Sie definierten kurze variable Segmente innerhalb von gyrB und rpoB, die sich mit standardmäßiger PCR amplifizieren und mit einem kuratierten Referenzsatz vergleichen lassen. Mit einer schrittweisen „B&B“-Strategie — zuerst gyrB, dann bei Bedarf rpoB — konnten sie Arten für 45 klinisch relevante Stämme mit perfekter Übereinstimmung zu den Ganzgenom‑ANI-Ergebnissen zuordnen, darunter Isolate aus kolorektalem Krebs und dem Mundraum.
Krebs‑Mikrobiomstudien mit besseren Bezeichnungen neu lesen
Um zu zeigen, warum korrekte Benennungen wichtig sind, integrierte das Team die revidierte Taxonomie in gängige genomische und metagenomische Tools wie die Genome Taxonomy Database und MetaPhlAn, die viele Gruppen zur Profilierung von Mikrobiomen aus Stuhl‑ oder Gewebeproben nutzen. Durch das Remappen von species‑level genome bins (SGBs), die in großen Studien zum kolorektalen Krebs verwendet wurden, entdeckten sie, dass mehrere bakterielle Signale, die ursprünglich allgemein als „F. nucleatum“ berichtet wurden, tatsächlich zu unterschiedlichen Arten gehören — etwa F. animalis, F. vincentii, F. polymorphum, F. nucleatum sensu stricto und F. watanabei. Diese feinere Auflösung ermöglicht es Forschern zu untersuchen, welche genaueren Arten in Tumoren angereichert sind, welche gesundes Gewebe besiedeln und wie sich ihre Rollen bei Entzündungen, Immunantworten oder Therapie‑Resistenz unterscheiden.
Was das für zukünftige Forschung und Versorgung bedeutet
Praktisch betrachtet ersetzt diese Arbeit ein unscharfes Bild von „F. nucleatum“ durch eine präzise Karte von 34 genetisch unterscheidbaren Fusobacterium‑Arten und bietet einen einfachen Zwei‑Gen‑Test, um sie ohne vollständige Genomsequenzierung auseinanderzuhalten. Für Klinikpersonal und Forschende bedeutet das klarere Verbindungen zwischen bestimmten Mikroben und konkreten Krankheitsausgängen, verlässlichere Vergleiche zwischen Studien und bessere Möglichkeiten, Diagnostik oder Therapien gegen Fusobacterium gezielt anzusetzen. Wenn in Zukunft weitere Genome hinzukommen, kann die genaue Grenze weiter verfeinert werden; das hier etablierte Rahmenwerk — basierend auf einer natürlichen genomischen Lücke und robusten Markergenen — liefert jedoch die taxonomische Präzision, die nötig ist, um sowohl die Grundlagenforschung als auch die klinische Forschung darüber voranzubringen, wie diese Mikroben Infektionen, Krebs und darüber hinaus beeinflussen.
Zitation: Bi, D., Wu, Y., Ji, G. et al. Integrating ANI and phylogenies for re-evaluation of Fusobacterium taxonomy and disease associations. Nat Commun 17, 3774 (2026). https://doi.org/10.1038/s41467-026-70540-x
Schlüsselwörter: Fusobacterium-Taxonomie, Mikrobiom und Krebs, Identifizierung bakterieller Arten, genombasierte Klassifikation, Mikrobiota des kolorektalen Karzinoms