Clear Sky Science · en
Cyclophilin A stabilizes the capsid protein p72 to facilitate African swine fever virus replication
Why a Pig Virus Matters to All of Us
African swine fever is a lethal disease of pigs that has devastated farms and food supplies across the world. With no effective vaccine or treatment available, understanding exactly how this virus multiplies inside cells is crucial for protecting global pork production and food security. This study uncovers how a common protein in pig cells helps the virus build its protective shell, and shows that blocking this partnership can sharply reduce viral growth.
How the Virus Builds Its Protective Shell
African swine fever virus (ASFV) is a large DNA virus wrapped in a hard outer shell, or capsid, mainly made of a protein called p72. This protein makes up about one-third of the virus’s mass and is essential for forming proper virus particles. Yet p72 is naturally unstable: cells tend to tag it for destruction, and earlier work showed it must be carefully dismantled when the virus first invades, then rebuilt later so new particles can form. The authors set out to find which pig cell proteins bind to p72 and might help protect it during this late stage when the virus is assembling.
The Host Helper: Cyclophilin A Steps In
Using infected pig immune cells, the team pulled down p72 and examined which host proteins came along for the ride. Mass spectrometry identified cyclophilin A (CypA), a very common protein in animal cells, as a strong binding partner. Follow-up experiments in both test tubes and living cells confirmed that CypA and p72 stick together directly and gather in the same spots within infected cells, especially in the so‑called viral factories where new virus particles are built. Structural modeling and mutational tests revealed that p72 docks onto a specific hollow surface on CypA, and changes that disrupt this cavity or block it with antibodies largely prevent p72 from binding.
Shielding the Capsid Protein from Destruction
The researchers then asked what this embrace between CypA and p72 actually does. They found that p72 is normally broken down mainly by the cell’s protein‑shredding machinery known as the proteasome, after being decorated with chains of small "ubiquitin" tags. When CypA was present, p72 protein levels rose, even though its gene activity did not change, indicating that CypA was stabilizing the protein rather than making more of it. Detailed assays showed that CypA sharply reduced a specific type of ubiquitin tagging on p72 that drives it toward destruction. In other words, when CypA binds p72, it acts as a shield that keeps the viral building block from being marked and fed into the cellular grinder.
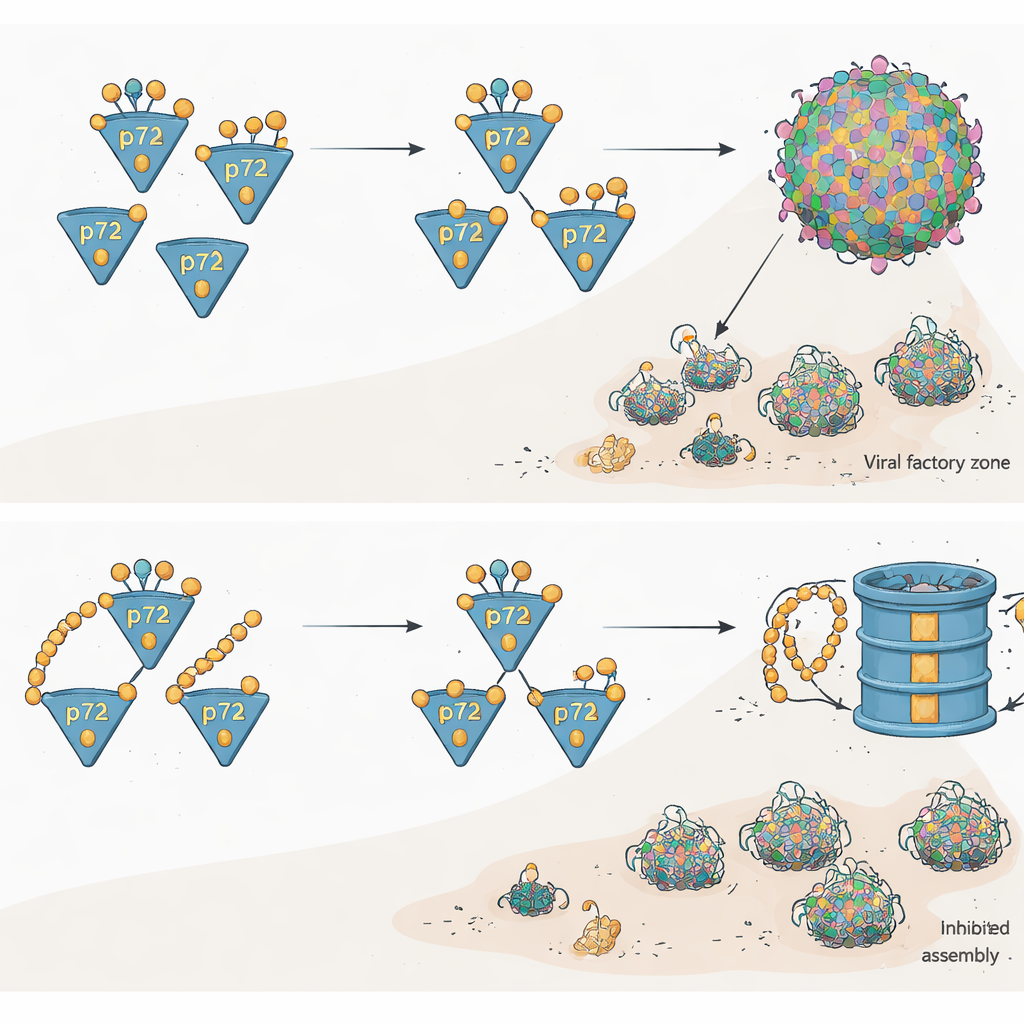
Turning the Shield into a Weakness
Because CypA is a known drug target, the team tested several small cyclic compounds that lodge inside the same cavity CypA uses to grip p72. These molecules disrupted the CypA–p72 interaction, increased ubiquitin tagging on p72, and sped up its breakdown. When pig cells or pig macrophages were treated with these inhibitors and then infected with ASFV, the virus struggled: key viral proteins—including p72 itself—built up much more slowly, viral factories were smaller and disorganized, and far fewer properly formed virus particles were seen under the electron microscope. Importantly, these effects were not due to the drugs killing the cells, and genetic removal of CypA produced similar defects in viral growth.
What This Means for Controlling the Disease
This work shows that ASFV relies on a host protein, cyclophilin A, to keep its main shell protein p72 from being destroyed inside infected cells. By protecting p72, CypA allows the virus to efficiently assemble new particles and spread. When CypA is blocked—either by gene editing or by specific drugs—p72 levels drop, viral factories malfunction, and the virus cannot replicate well. For a lay reader, the take‑home message is that the virus has a critical "accomplice" inside pig cells, and carefully designed CypA‑targeting drugs could turn that accomplice into a powerful point of attack for future antiviral strategies against African swine fever.
Citation: Kong, H., Yang, L., Zhang, Z. et al. Cyclophilin A stabilizes the capsid protein p72 to facilitate African swine fever virus replication. Nat Commun 17, 3624 (2026). https://doi.org/10.1038/s41467-026-70430-2
Keywords: African swine fever virus, capsid protein p72, cyclophilin A, host–virus interaction, antiviral drug targets