Clear Sky Science · en
GPR146 in adipose tissue drives adipose-liver crosstalk and promotes hepatic steatosis in mice
Why this matters for everyday health
Many people carry extra weight and have fatty liver without knowing it. This silent buildup of fat in the liver can lead to serious problems, including liver failure and cancer, yet treatment options remain limited. This study uncovers a hidden communication line between body fat and the liver, controlled by a little-known cell-surface switch called GPR146. Understanding how this switch works could open the door to new medicines that protect the liver by acting on fat tissue, rather than the liver itself.
A quiet epidemic in the liver
Metabolic dysfunction-associated steatotic liver disease (MASLD), formerly grouped under nonalcoholic fatty liver disease, affects about one in four adults worldwide. In its early form, the liver fills with fat; in more severe stages, called MASH, long-term inflammation and scarring can progress to cirrhosis and liver cancer. Currently, only a couple of drugs are approved to treat these conditions, so researchers are urgently looking for new biological pathways that could be targeted safely. One strong suspect is the fat tissue itself: when overloaded with nutrients, it releases a flood of free fatty acids and inflammatory signals into the bloodstream, which then burden the liver. This study asks whether GPR146, a receptor found on cell surfaces, helps drive that harmful fat–liver traffic.
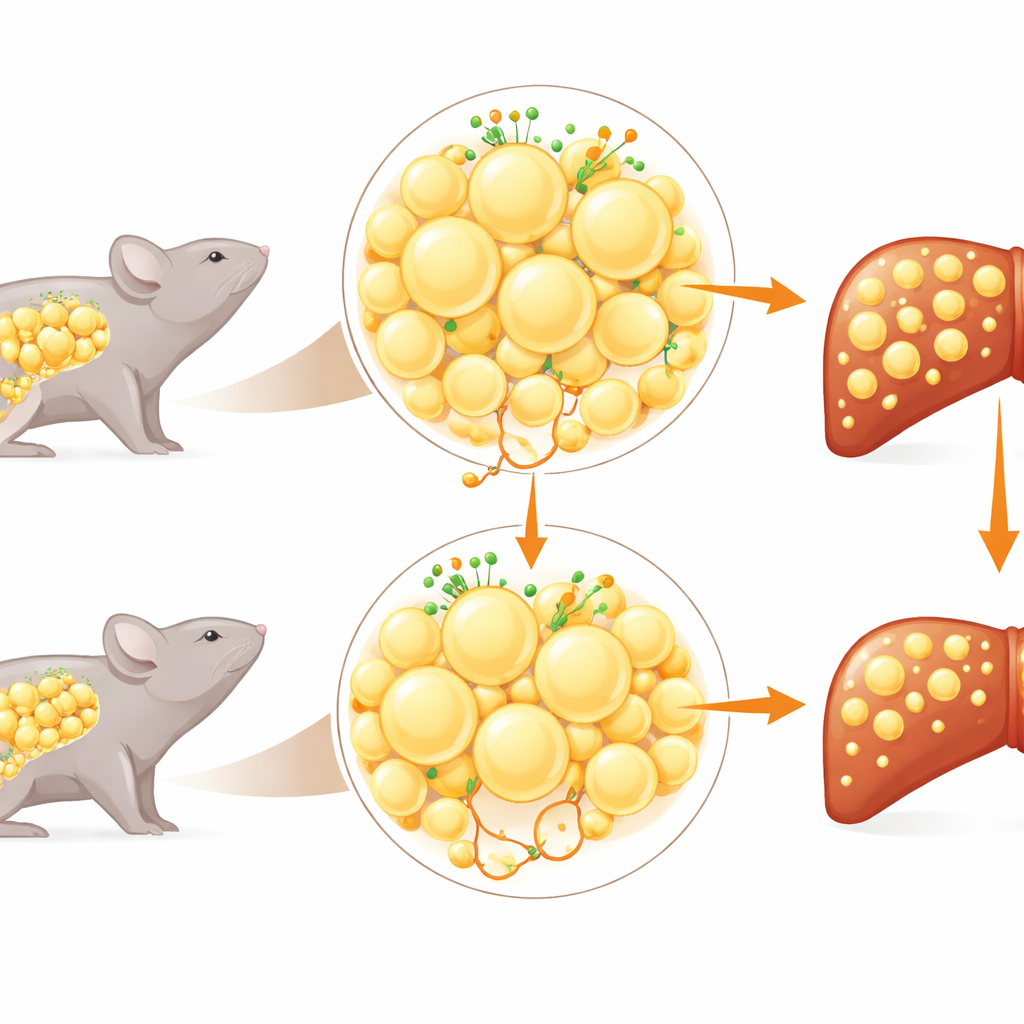
A fat-tissue switch with a liver impact
GPR146 was first linked to blood cholesterol levels in large human genetics studies. Here, the authors show that the same DNA region is also associated with blood markers of liver injury and inflammation. They then turned to mouse models to probe cause and effect. Mice lacking GPR146 throughout the body were fed high-fat, liver-stressing diets. Compared with normal mice, the knockouts gained less weight, had smaller fat cells, and showed much less fat and inflammation in their livers. Chemical profiling of liver tissue revealed lower levels of multiple fat species and damaging oxidized lipids, along with reduced activation of genes tied to scarring and immune responses. At the same time, these animals showed signs of using more sugar for fuel and storing more glycogen, hinting at a healthier way of handling nutrients overall.
Fat talks to liver, not the other way around
A key question was where GPR146’s most important actions occur. Surprisingly, deleting the receptor only in the liver did not improve fatty liver; in some cases liver weight even increased. In contrast, deleting GPR146 only in fat tissue lowered body weight, shrank white fat depots, reduced free fatty acids in the blood, and markedly cut liver fat and scarring-related gene activity. An independent approach using a virus to acutely silence GPR146 in adult mice—mainly in fat depots and liver—confirmed these protective effects. Together, these results show that it is GPR146 in fat tissue, rather than in the liver itself, that drives the excess flow of fat from adipose tissue to the liver.
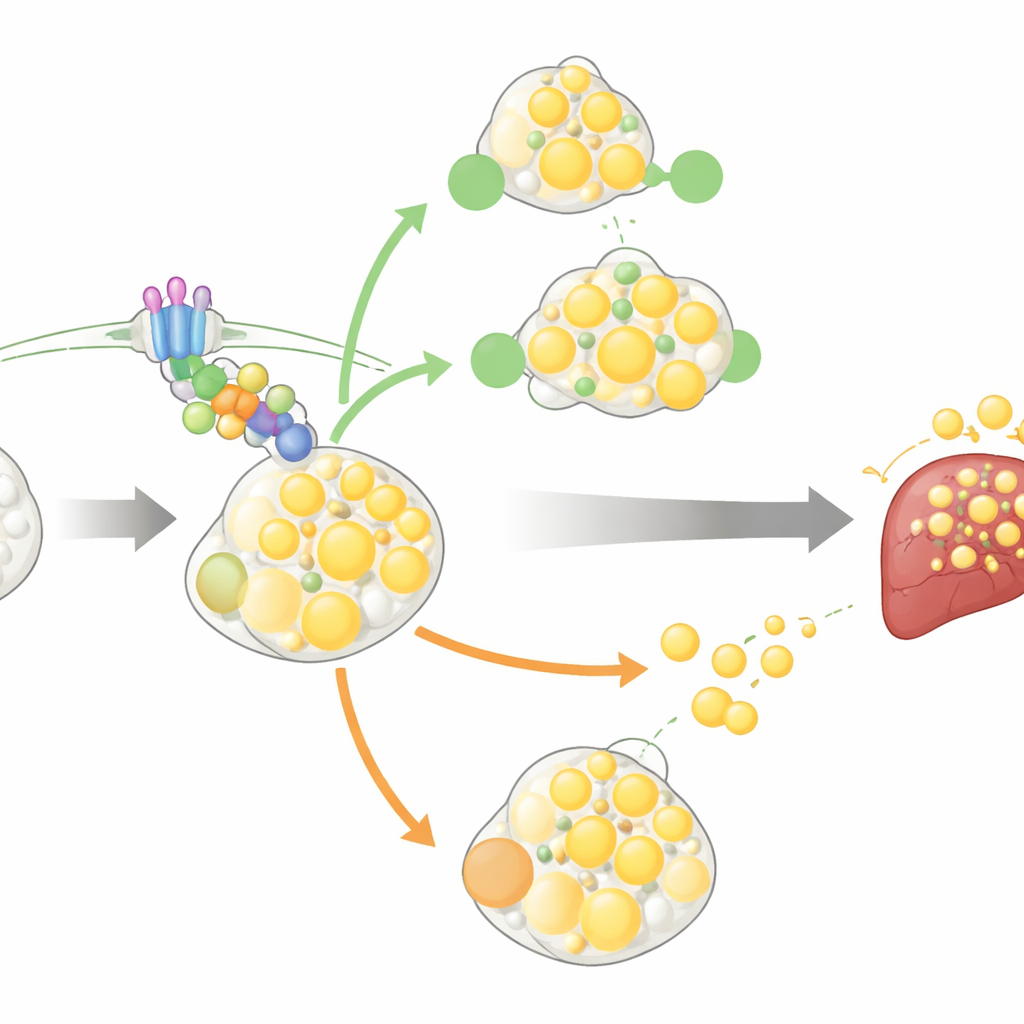
How the switch changes fat cells
To understand the mechanism, the researchers studied both mouse and human pre-fat cells in the lab. As these cells matured into fat-storing adipocytes, GPR146 levels naturally rose. When the receptor was knocked down, fewer cells completed the transition into fully loaded fat cells, and those that did contained less fat. This was traced to weakened activity in a signaling chain that runs through a G-protein (called Gαq), protein kinase C (PKC), and the AKT pathway—signals known to support cell growth and filling with lipid. Blocking PKC erased the difference between normal and GPR146-deficient cells, underscoring that this route is essential for the receptor’s effect on fat-cell formation. Interestingly, too much GPR146 also disrupted differentiation, suggesting that its activity must be finely tuned for fat tissue to expand normally.
A two-faced role in mature fat cells
GPR146 does not stop working once fat cells are mature. In fully developed human adipocytes, dialing down GPR146 reduced the breakdown of stored fat, while boosting it increased fat release. Here, the key pathway involved was ERK signaling rather than the earlier PKC–AKT route. In mice with GPR146 knocked down in adulthood, stimulation of fat breakdown in vivo also produced a weaker rise in blood glycerol, a marker of lipolysis. This means GPR146 helps both to build up fat stores and to liberate fat from those stores into the circulation. Under calorie excess, those dual actions add up: more fat cells, more fat turnover, and more free fatty acids reaching the liver, where they accumulate as triglycerides.
What this could mean for future therapies
By mapping how GPR146 governs the back-and-forth of fat between adipose tissue and liver, this work highlights a new target for treating fatty liver disease and obesity. Turning down this receptor specifically in fat tissue in mice makes them leaner, less inflamed, and far less prone to fatty liver, without obvious harm to other organs. The study also reveals sex-specific differences in how energy is burned, hinting that treatments may need to be tailored for men and women. While many steps remain before new drugs reach patients, GPR146 now stands out as a promising lever for rebalancing how the body stores and ships fat—and for easing the hidden burden of fatty liver disease.
Citation: Shi, Y., Cheng, K.Y., Thi, T.T. et al. GPR146 in adipose tissue drives adipose-liver crosstalk and promotes hepatic steatosis in mice. Nat Commun 17, 3389 (2026). https://doi.org/10.1038/s41467-026-70136-5
Keywords: fatty liver disease, adipose tissue, G protein-coupled receptors, lipid metabolism, metabolic syndrome