Clear Sky Science · en
Targeting oncogene-induced senescence in ETV6::RUNX1 pre-leukemic cells
Hidden Seeds of Childhood Leukemia
Most children who develop leukemia are diagnosed only after the disease is in full swing. Yet for many, the story starts years earlier, with a small population of altered blood cells quietly lingering in the body. This study explores why those “pre-leukemic” cells can survive for so long, how they may later fuel cancer, and whether we can eliminate them before they cause harm—offering a glimpse of truly preventive cancer therapy.
A Quiet Alteration Before Birth
In some newborns, a tiny genetic reshuffling joins two genes, ETV6 and RUNX1, into a single hybrid. This ETV6::RUNX1 fusion shows up in the blood of 2–5% of healthy babies, but only about 1% of these children ever go on to develop a common form of childhood blood cancer called B‑cell precursor acute lymphoblastic leukemia. The fusion gene creates abnormal early B‑cell precursors that partially mature and then stall. They can persist for years without causing symptoms, forming a hidden reservoir of pre-leukemic cells that may later acquire additional mutations and transform into full-blown leukemia.
When a Cancer Gene Makes Cells Grow Old
Instead of simply driving uncontrolled growth, the ETV6::RUNX1 fusion pushes cells into a senescence-like state—something akin to cellular old age. Using a mouse pro‑B cell line and a transgenic mouse model, the researchers found that cells expressing the fusion became larger, flatter, and slowed their cell cycle. They showed high activity of a classic aging marker enzyme, produced excess reactive oxygen species, and secreted a stew of inflammatory molecules known as the senescence-associated secretory phenotype. These changes mirror a process called oncogene-induced senescence, in which an early cancer-causing event paradoxically forces cells to pause growth.
A Bent Safety Switch in the Cell
Normally, when cells accumulate DNA damage or experience strong oncogenic signals, a guardian protein called p53 helps decide their fate—either stop dividing or self-destruct. In the ETV6::RUNX1‑positive cells, gene activity patterns showed that p53 was strongly engaged in enforcing cell-cycle arrest but not in turning on many of its usual death-promoting partners. The cells built up p53 protein and activated stress-related signaling, yet one crucial modification of p53—a phosphate tag at a specific site needed for efficient apoptosis—was missing. This defect was linked to reduced levels of a kinase enzyme that normally adds this tag. As a result, when the cells were exposed to DNA-damaging treatments such as chemotherapy-like drugs or radiation, they were more likely to survive than their normal counterparts.
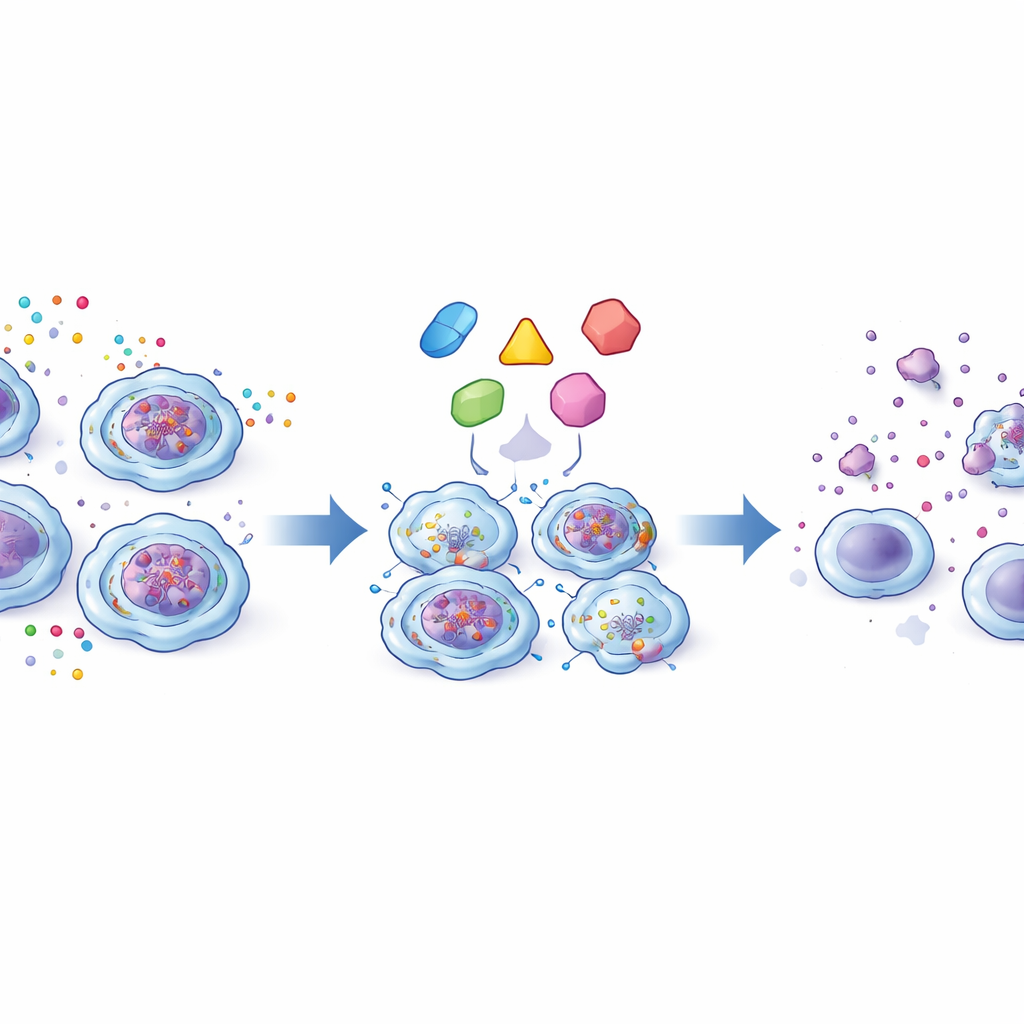
Turning Senescence into a Therapeutic Target
Seeing that ETV6::RUNX1‑positive cells were senescent yet stubbornly alive, the team tested whether they could be selectively removed using senolytic drugs, which are designed to kill senescent cells. One compound, SSK1, is activated by the same beta-galactosidase enzyme that is overactive in these cells and released a toxic payload only inside them. Another, piperlongumine—a natural product from long pepper—exploited their already high oxidative stress, pushing them over the edge into cell death while sparing normal cells. A third, TM5441, blocked PAI‑1, a protein that helps senescent cells resist apoptosis. All three agents preferentially killed fusion‑positive cells in culture. In the transgenic mice, treating bone marrow progenitors with SSK1 reduced the formation of pre‑B cell colonies derived from ETV6::RUNX1‑expressing cells, but not from normal cells, reinforcing the idea that senescence-linked features can be used as a therapeutic handle.
From Early Checkpoint to Prevention Strategy
This work reframes the ETV6::RUNX1 fusion not as a simple on‑switch for leukemia, but as a trigger for a strange in‑between state: cells that have stopped dividing, resist dying, and quietly accumulate damage over time. That pre-leukemic checkpoint may help explain why only a small fraction of carriers eventually develop cancer. At the same time, it exposes specific weaknesses—from enzyme activities to stress defenses—that senolytic drugs can exploit. In the future, carefully targeted treatments might one day clear these lingering pre-leukemic cells in high‑risk individuals, reducing relapse after therapy or even preventing leukemia before it starts.
Citation: Acunzo, D., Bertagna, M., Risca, G. et al. Targeting oncogene-induced senescence in ETV6::RUNX1 pre-leukemic cells. Cell Death Discov. 12, 145 (2026). https://doi.org/10.1038/s41420-026-03001-5
Keywords: childhood leukemia, pre-leukemic cells, cellular senescence, ETV6-RUNX1 fusion, senolytic therapy