Clear Sky Science · en
PABPC1-induced stabilization of PGK1 mRNA reduces apoptosis and sunitinib sensitivity in renal cell carcinoma by suppressing endoplasmic reticulum stress
Why kidney cancer drug resistance matters
Many people with advanced kidney cancer rely on targeted drugs to keep their disease in check. One of the most widely used medicines, sunitinib, often works at first but then loses its punch as tumors become resistant. This study explores why some kidney tumors stop responding and points to a possible way to make sunitinib effective again.
A closer look at stubborn kidney tumors
The researchers focused on clear cell renal cell carcinoma, the most common form of kidney cancer. Using large patient data sets and tumor samples, they found that a molecule called PABPC1 was present at higher levels in cancer tissue than in normal kidney tissue. Patients whose tumors had more PABPC1 tended to have more aggressive disease, more spread to lymph nodes and distant sites, and shorter survival. In cell culture, dialing down PABPC1 slowed cancer cell growth and movement, while boosting it made the cells grow and invade more readily.
How tumor cells dodge sunitinib
Because sunitinib is a key first-line treatment, the team asked whether PABPC1 is involved in resistance to this drug. They compared standard kidney cancer cells with cells that had been made resistant to sunitinib in the lab, as well as tumor samples from patients who had stopped responding to treatment. In all of these resistant cells and tissues, PABPC1 levels were higher. When the researchers reduced PABPC1, the cells became easier to kill with sunitinib and showed more signs of programmed cell death. In mice, tumors with lower PABPC1 grew more slowly, especially when treated with sunitinib, and showed fewer dividing cells and more dying cells.
A hidden stress system inside cancer cells
The study then turned to a key quality-control system inside cells called endoplasmic reticulum, or ER, stress. This system reacts when proteins become misfolded and can push damaged cells toward self-destruction. The scientists found that lowering PABPC1 switched on ER stress in resistant kidney cancer cells, while raising PABPC1 shut it down. Under the microscope, cells lacking PABPC1 showed swollen and disrupted ER structures, and key ER stress sensors were more active. Blocking ER stress with a chemical inhibitor erased much of the benefit seen when PABPC1 was reduced, suggesting that this stress pathway is central to how the tumors respond to the drug.
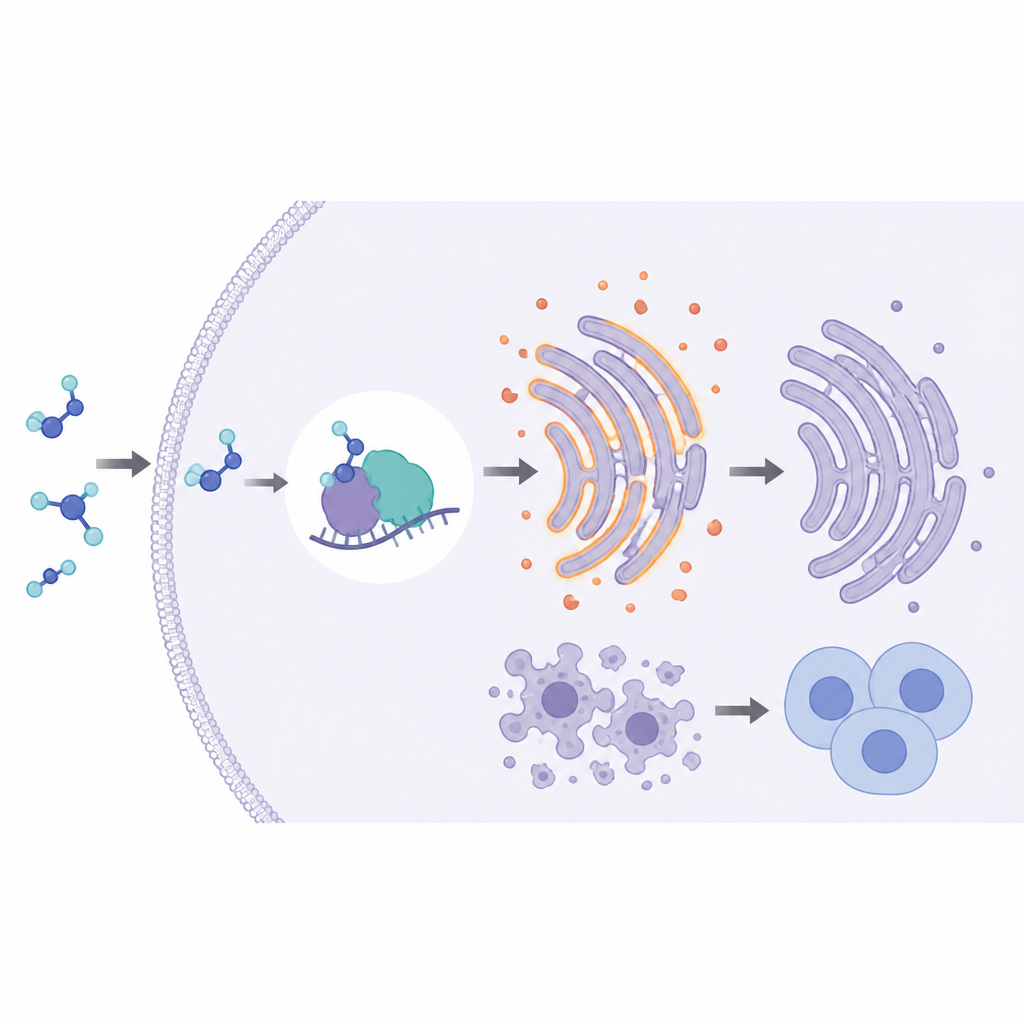
The PABPC1–PGK1 partnership
To understand how PABPC1 controls ER stress, the team searched for partner molecules whose levels dropped when PABPC1 was silenced. One enzyme, PGK1, stood out. PGK1 helps cancer cells fuel their growth through altered sugar metabolism and was especially abundant in sunitinib-resistant cells and patient tumors. The researchers showed that PABPC1 physically binds to the tail region of PGK1’s genetic message and keeps it from being broken down, thereby raising PGK1 levels. When PGK1 was restored in cells lacking PABPC1, the tumors regained their fast growth, ER stress faded, and resistance to sunitinib returned in both cell cultures and mouse models. This placed PGK1 as a key middleman linking PABPC1 to drug resistance.
A possible way to restore drug sensitivity
Finally, the investigators tested whether deliberately turning ER stress back on could overcome resistance. They used a small molecule called Eeyarestatin I, which disrupts protein handling in the ER and heightens stress levels. On its own, it slowed tumor growth, but when combined with sunitinib it shrank tumors in mice far more than either treatment alone, without clear signs of added toxicity. The combination lowered blood vessel growth in tumors and increased cell death markers, indicating a stronger anticancer effect even in settings where PABPC1 and PGK1 were high.
What this means for patients
In simple terms, this work shows that some kidney tumors use the PABPC1–PGK1 pair to silence an internal stress alarm that would otherwise help sunitinib kill cancer cells. By stabilizing PGK1, PABPC1 calms this stress system, reduces cell death, and makes the drug less effective. Reactivating ER stress with agents like Eeyarestatin I restored the tumors’ vulnerability to sunitinib in the lab and in mice. While more research is needed before this strategy reaches the clinic, the study maps out a clear molecular route that future therapies could target to tackle drug-resistant kidney cancer.
Citation: Chen, X., Cao, S., Jia, T. et al. PABPC1-induced stabilization of PGK1 mRNA reduces apoptosis and sunitinib sensitivity in renal cell carcinoma by suppressing endoplasmic reticulum stress. Cell Death Dis 17, 452 (2026). https://doi.org/10.1038/s41419-026-08676-3
Keywords: kidney cancer, sunitinib resistance, endoplasmic reticulum stress, RNA binding proteins, PGK1