Clear Sky Science · de
Hochauflösende Kartierung von Proteinbewegungen in Zeit und Raum mit RMSX und Flipbook
Proteine in Bewegung beobachten
Proteine in unseren Zellen sind keine starren Skulpturen; sie drehen sich, biegen sich und „atmen“, während sie ihre Aufgaben erfüllen. Viele wichtige biologische Prozesse – von der Reifung von Viren bis zum Festhalten von Bakterien an unserem Gewebe – hängen davon ab, wann und wo sich Teile eines Proteins genau bewegen. Die meisten Computerwerkzeuge zeigen jedoch entweder die mittlere Bewegung über die Zeit oder die Gesamtveränderung der Form, wodurch kurzlebige, lokale Bewegungen schwer zu erkennen sind. Dieser Artikel stellt zwei neue Methoden vor, RMSX und Flipbook, die komplexe Simulationsdaten in klare, detaillierte Darstellungen von Proteinbewegungen in Zeit und Raum verwandeln und es Forschern erleichtern, wichtige molekulare Ereignisse zu entdecken und zu erklären.
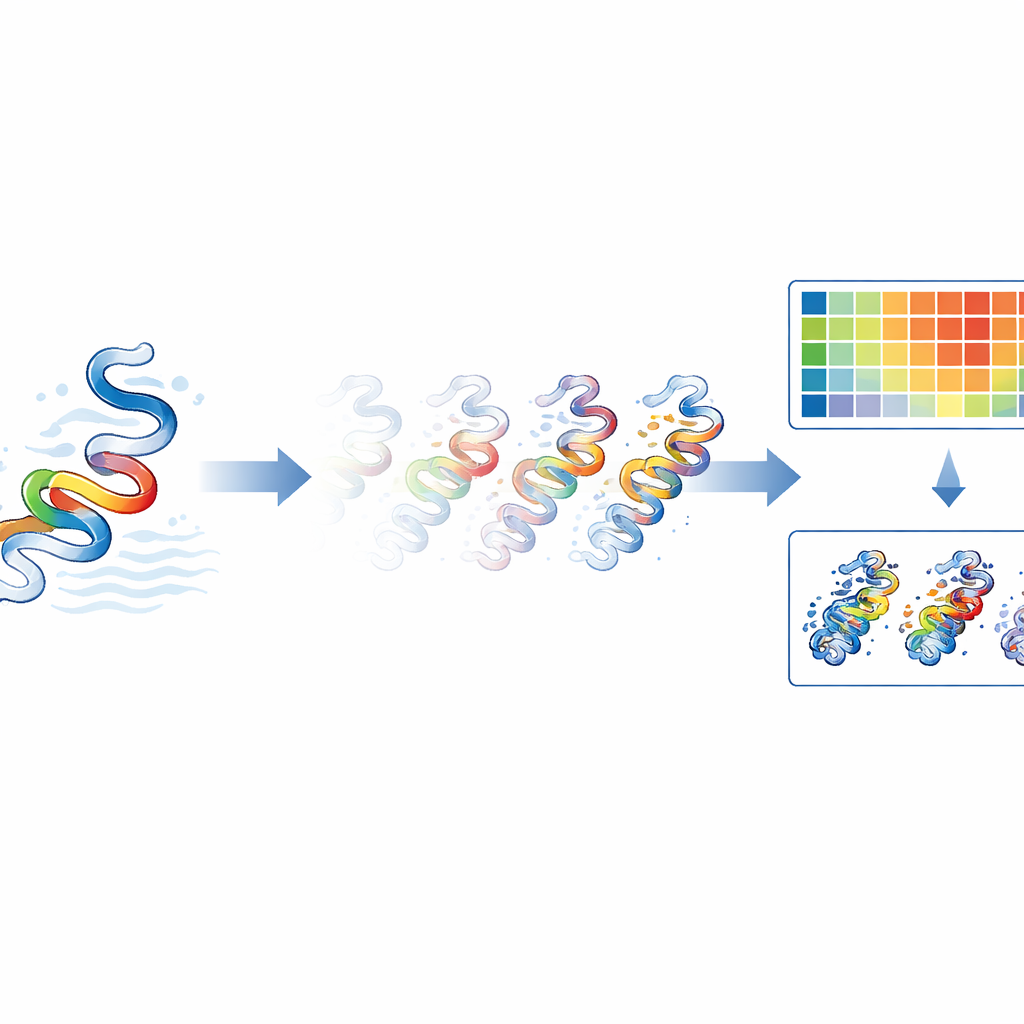
Eine neue Methode, zappelnde Teile zu verfolgen
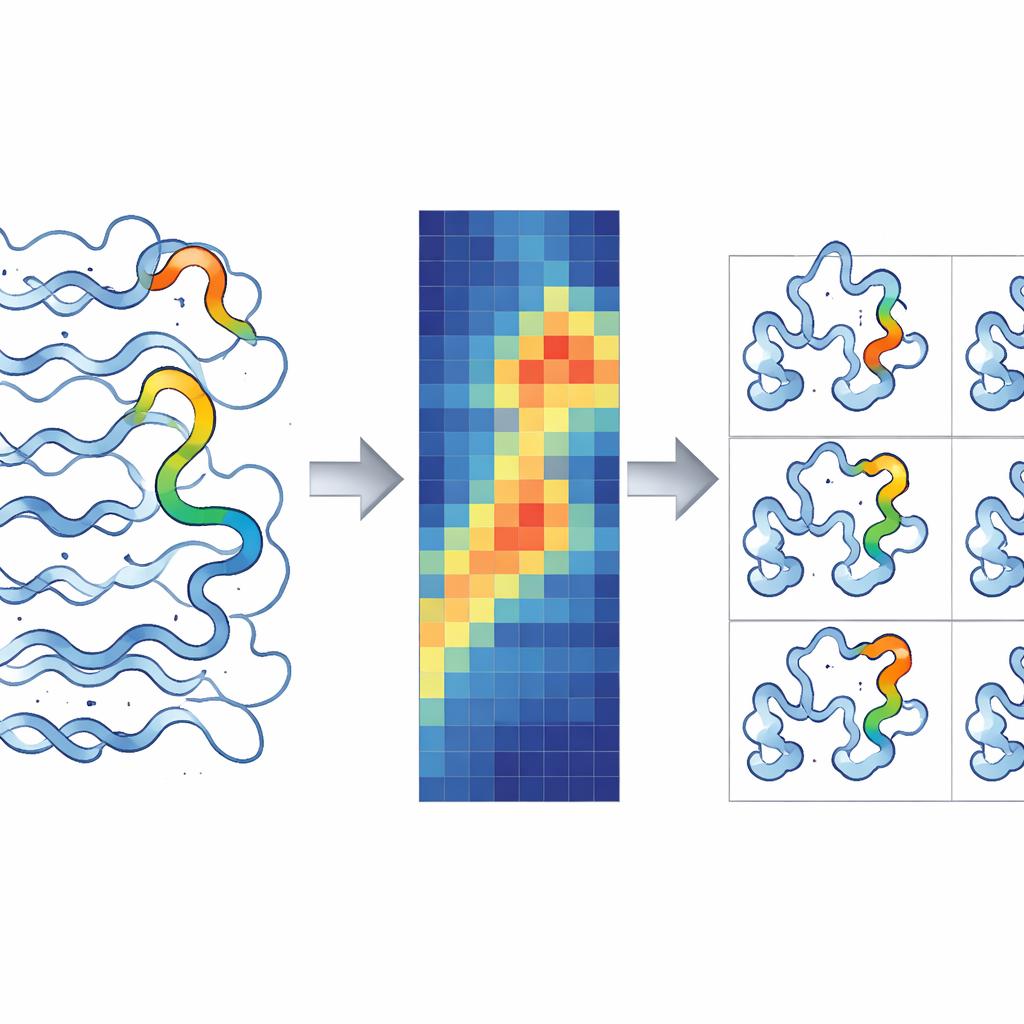
Traditionelle Größen in molekularen Simulationen wie Root Mean Square Deviation (RMSD) und Root Mean Square Fluctuation (RMSF) liefern nur einen Teil der Geschichte. RMSD sagt aus, wie weit die Gesamtform eines Proteins von seiner Ausgangsstruktur abweicht, während RMSF beschreibt, wie viel sich jede Aminosäure im Durchschnitt während der gesamten Simulation bewegt. Keine dieser Größen kann für eine bestimmte Restposition sowohl angeben, wie stark sie sich bewegt, als auch genau wann diese Bewegung stattfindet. RMSX löst dies, indem es eine Simulation in Zeitfenster unterteilt und für jedes Fenster die Bewegung pro Rest berechnet. Die Ergebnisse werden zu einer Heatmap zusammengesetzt, bei der eine Achse die Position im Protein, die andere die Zeit repräsentiert und die Farben zeigen, wie stark jeder Teil des Proteins zu jedem Moment schwankt. Diese einfache Umordnung bekannter Berechnungen liefert eine hochauflösende Sicht auf verschiebende Proteinregionen, die sonst übersehen würden.
Zahlen in bewegte Bilder verwandeln
Während RMSX reichhaltige numerische Daten erzeugt, müssen Wissenschaftler diese Bewegungen am tatsächlichen 3D-Modell sehen. Genau dafür ist Flipbook konzipiert. Es nimmt Werte wie RMSX oder andere pro-Rest-Metriken und kodiert sie in standardmäßige Proteinstrukturdateien, wie sie gängige Molekularviewer verstehen. Werden diese Schnappschüsse in Werkzeuge wie ChimeraX oder VMD geladen, kann jede Aminosäure je nach Bewegung eingefärbt und dicker dargestellt werden, und die Schnappschüsse werden in einer Reihenfolge wie Frames eines Daumenkino abgelegt. Das Ergebnis ist ein visuelles „Flipbook“, das es Betrachtern erlaubt, zu verfolgen, wie sich bestimmte Schleifen oder Segmente im Laufe der Zeit wiegen, dehnen oder starr bleiben. Da dieselben Farbskalen für Heatmaps und 3D-Ansichten verwendet werden, ist es einfach, einen hellen Fleck in einem Diagramm mit der genauen Region des Proteins zu verbinden, die sich bemerkenswert verhält oder aktiv wird.
Die Werkzeuge an realen molekularen Beispielen testen
Um zu demonstrieren, was diese Werkzeuge aufdecken können, wendeten die Autoren RMSX und Flipbook auf drei sehr unterschiedliche Proteine an. In einer erzwungenen Entfaltungssimulation von Ubiquitin – einem kleinen, federnden Protein – zeigten sie, wie sich die Bewegung an den Kettenenden konzentriert, während ein fixierter Ankerpunkt unbeweglich bleibt. Flipbook macht diese Entfaltung sichtbar wie eine auseinandergezogene Feder, bei der ausgewählte Reste zu bestimmten Zeiten wegschwingen. Beim HIV-1-Protease, einem Schlüsselenzym im Lebenszyklus von HIV, lag der Fokus auf zwei flexiblen „Klappen“, die sich öffnen und schließen, um Wirkstoffmoleküle oder natürliche Substrate hereinzulassen. RMSX-Heatmaps und Flipbook-Ansichten hoben deutlich die Spitzen dieser Klappen hervor und zeigten ruhige Intervalle, in denen sie geschlossen bleiben, sowie dynamische Perioden mit vorübergehendem Öffnen – Details, die durch Medikamentenresistenzmutationen verändert werden können.
Erkennen, wie Proteine Kräften widerstehen
Der dritte Testfall betraf SdrG, ein bakterielles Adhäsionsprotein, das mit außergewöhnlicher Kraft an menschliches Fibrinogen bindet. Unter starkem Zug werden Teile von SdrG straffer und verändern ihre Lage so, dass die Bindung tatsächlich stabilisiert wird – ein Phänomen, das als Catch-Bond bezeichnet wird. Durch die Kombination von RMSX mit einer weiteren Metrik, die kumulative Verschiebungen über die Zeit verfolgt, und durch Visualisierung beider Kenngrößen mit Flipbook konnten die Autoren beobachten, wie bestimmte Schleifen sich straffen, umordnen und dann allmählich entspannen, während der Zug anhält. Diese Paarung erlaubte es ihnen, einfachen Drift des Proteins von echten, kurzzeitigen lokalen Bewegungsstößen zu trennen und so ein vollständigeres Bild davon zu zeichnen, wie mechanische Kraft die Bindungsstelle umgestaltet.
Was das für die Proteinwissenschaft bedeutet
Schließlich bieten RMSX und Flipbook ein praktisches, Open-Source-Toolkit, um rohe Simulationsverläufe in klare, publikationsreife Erzählungen über Proteinbewegungen zu verwandeln. RMSX vereint die Stärken älterer Maße, indem es in einer einzigen Ansicht zeigt, welche Reste sich bewegen und wann dies geschieht. Flipbook projiziert diese Zahlen anschließend auf 3D-Strukturen und verwandelt abstrakte Kurven und Raster in intuitive Szenen von biegenden Schleifen und starren Kernen. Zusammen mit anderen Maßen, die langfristigen Drift oder feine lokale Umordnungen verfolgen, helfen diese Werkzeuge Forschern, flüchtige strukturelle Ereignisse zu entdecken, die Allosterie, Kraftwahrnehmung oder Wirkstoffbindung zugrunde liegen können. Für Nicht-Fachleute bieten sie zudem eine zugänglichere Möglichkeit, das unruhige Leben der Proteine, die die Biologie antreiben, „zu sehen“.
Zitation: Beruldsen, F., de Freitas, M.V. & Antunes, D.A. High resolution mapping of protein motions in time and space with RMSX and Flipbook. Sci Rep 16, 10035 (2026). https://doi.org/10.1038/s41598-026-39869-7
Schlüsselwörter: Protein-Dynamik, molekulare Simulationen, Bewegungsvisualisierung, Proteinflexibilität, biomolekulare Struktur