Clear Sky Science · de
Chromosomenebenen-Genom der Rötelmaus (Clethrionomys glareolus): eine Ressource für Eco-Evo-Disease-Forschung
Ein kleines Waldbewohner mit großem wissenschaftlichem Potenzial
Stellen Sie sich ein gewöhnliches Waldnagetier vor, das leise dabei hilft, Fragen zu Klimawandel, neu auftretenden Krankheiten und der Anpassung des Lebens im Lauf der Zeit zu beantworten. Dieses Tier ist die Rötelmaus, ein kleines, mausähnliches Säugetier, das in ganz Europa und Westasien vorkommt. In dieser Studie haben Forschende die DNA der Rötelmaus zu einer vollständigen, chromosomenebenen Karte zusammengesetzt. Dieser detaillierte genetische Bauplan macht das unscheinbare Waldtier zu einer leistungsfähigen Referenzart für Studien von Evolution und Ökologie bis hin zu Virusinfektionen und Immunabwehr.
Warum die DNA eines kleinen Nagetiers kartieren?
Jahrzehntelang konzentrierte sich die meiste genetische Forschung auf einige klassische „Labormodelle“ wie Mäuse und Ratten, die eher aus praktischen Gründen als wegen ökologischer Relevanz gewählt wurden. Sinkende Kosten für DNA-Sequenzierung haben dieses Bild verändert. Wissenschaftler können heute hochwertige Genome für Wildarten erstellen, die in realen, sich verändernden Umgebungen leben. Die Rötelmaus ist ein Paradebeispiel: sie ist weit verbreitet, klimatolerant empfindlich und in Bezug auf die Wanderungen und Anpassungen ihrer Populationen während und nach den Eiszeiten bereits gut untersucht. Außerdem trägt sie natürlicherweise mehrere durch Nagetiere übertragene Viren und Parasiten, was sie nützlich macht, um zu verstehen, wie Krankheitserreger in der Wildnis zirkulieren und gelegentlich auf den Menschen überspringen.
Erstellung eines vollständigen genetischen Bauplans
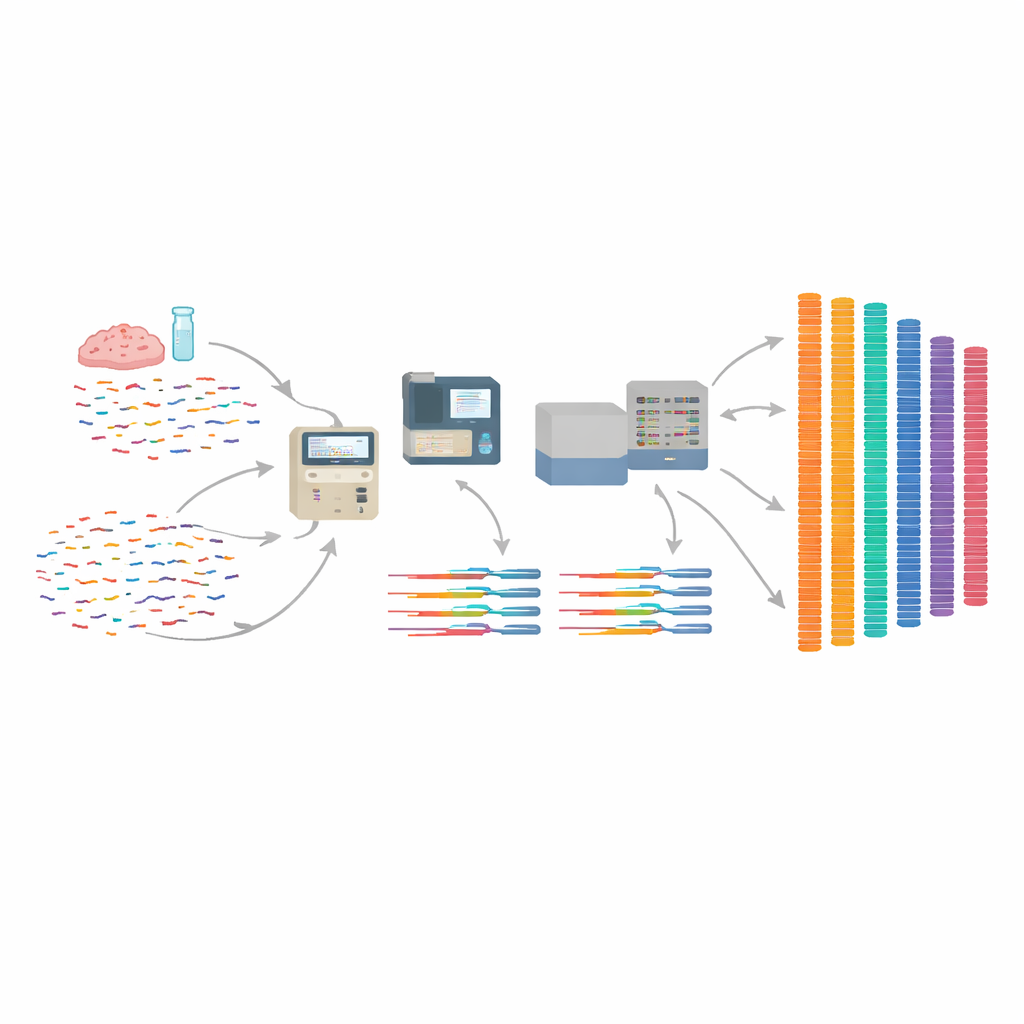
Das Team wollte frühere, fragmentierte Versionen des Rötelmausgenoms zu einer ordentlichen, chromosomenweisen Assembly aufwerten. Sie kombinierten große Mengen kurzer DNA-Lesungen aus Standardsequenzierung mit speziellen „Chicago“- und „Hi-C“-Daten, die erfassen, wie DNA-Stücke innerhalb der Zelle physisch verbunden sind. Mit einer rechnerischen Pipeline namens HiRise setzten sie Hunderttausende kleiner Fragmente zu nur 28 langen Scaffolds zusammen – jeweils entsprechend einem Chromosom und im Einklang mit den bereits bekannten Befunden aus mikroskopischen Untersuchungen der Rötelmaus-Chromosomen.
Qualitätsprüfung und Auffinden der Gene
Um zu prüfen, ob wichtige Abschnitte fehlen, nutzten die Forschenden eine weit verbreitete Qualitätskontrolle, die nach einem Kernsatz essenzieller Gene sucht, die bei Säugetieren erwartet werden. Die finale Assembly erfasste etwa 91 % dieser Benchmark-Gene, was auf ein hohes Maß an Vollständigkeit hinweist. Anschließend kombinierten sie Informationen aus Rötelmaus-RNA und einer sorgfältig kuratierten Proteindatenbank, um vorherzusagen, wo Gene im Genom liegen und welche Funktionen sie haben könnten. Sie identifizierten über 40.000 Genregionen, darunter mehr als 21.000 wahrscheinlich proteinkodierende Gene – Zahlen, die denen von Maus und Ratte sehr ähnlich sind. Die meisten dieser Gene ähneln stark den bereits aus den klassischen Laborroden bekannten Genen, was den Wissensübertrag zwischen den Arten erleichtern wird.
Aufdeckung repetitiver DNA und versteckter Fallstricke
Nicht alle DNA kodiert Gene. Ein großer Teil besteht aus sich wiederholenden Sequenzen und mobilen Elementen, die sich im Laufe der Evolution im Genom kopiert und eingefügt haben. Mit spezieller Software fanden die Autorinnen und Autoren heraus, dass etwa 29 % des Rötelmausgenoms aus solchen Wiederholungen bestehen, hauptsächlich aus zwei großen Klassen, die als LINEs und SINEs bekannt sind, sowie weiteren mobilen Elementen. Dieses Gesamtmuster stimmt eng mit dem verwandter Nagetierarten überein. Sie identifizierten außerdem Regionen, in denen kurze DNA-Lesungen schwer eindeutig zuzuordnen sind, und erstellten eine „maskierte“ Version des Genoms, die anderen Forschenden hilft, irreführende Signale zu vermeiden, wenn sie neue Daten auf diese Referenz abbilden.
Eine neue Grundlage für Klima- und Krankheitsforschung
Mit diesem Chromosomenebenen-Genom zur Hand wird die Rötelmaus zu einer deutlich stärkeren Modellart. Forschende können nun winzige DNA-Unterschiede mit Eigenschaften wie Kälteresistenz, Veränderungen der Körperfunktionen oder Infektionsresistenz verknüpfen und genau sehen, wo diese Unterschiede entlang der Chromosomen liegen. Die neue Referenz wird Studien unterstützen, wie Wildpopulationen auf sich verändernde Klimabedingungen reagieren, wie Krankheitserreger und Wirte ko-evolvieren und wie genetische Variation über Landschaften verteilt ist. Kurz gesagt: Dieses kleine Waldbewohner hat jetzt ein großes, gut organisiertes Bedienungsanleitungs-ähnliches Nachschlagewerk, und diese Ressource wird Forschenden helfen, drängende Fragen zu Biodiversität, Anpassung und Krankheitsausbreitung anzugehen.
Zitation: Marková, S., White, T.A., Searle, J.B. et al. Chromosome-level genome of the bank vole (Clethrionomys glareolus): a resource for eco-evo-disease research. Sci Data 13, 536 (2026). https://doi.org/10.1038/s41597-026-06924-x
Schlüsselwörter: Genom der Rötelmaus, Chromosomenebenen-Assembly, ökologisch-evolutionäre Genomik, durch Nagetiere übertragene Krankheiten, Anpassung an das Klima