Clear Sky Science · de
Chromosomenebene-Genomassemblierung von Chirolophis japonicus Herzenstein, 1890 (Stichaeidae, Perciformes)
Ein verborgener Bewohner kalter Felsküsten
Entlang der kühlen Küsten des nordwestlichen Pazifiks lebt Chirolophis japonicus, ein schlanker Fisch mit eigenartigen Fransen am Kopf, die ihm helfen, sich in Felsriffen zu tarnen. Obwohl er unscheinbar wirkt, spielt diese Art eine wichtige Rolle in küstennahen Nahrungsnetzen und steht unter Druck durch Überfischung, Verschmutzung und Lebensraumverlust. Um zu verstehen, wie dieser Fisch lebt, sich an kalte Gewässer anpasst und wie man ihn am besten schützt, haben Wissenschaftler nun seine DNA auf Chromosomenebene entschlüsselt und damit einen detaillierten genetischen Bauplan erstellt, der bisher fehlte.
Warum dieser Küstenfisch wichtig ist
Chirolophis japonicus verbringt sein Leben dicht am Meeresboden auf flachen Felsriffen, wo er sich von kleinen Fischen, Algen und Schalentieren ernährt. Er reift schnell, paart sich etwa im Alter von zwei Jahren und laicht im Herbst. In den letzten Jahrzehnten sind lokale Bestände in einigen Regionen jedoch zurückgegangen, was breitere Rückgänge in der Meeresfischerei widerspiegelt. Trotz seiner ökologischen Bedeutung und Verletzlichkeit gab es für diese Art kein hochwertiges Referenzgenom, was es schwer machte zu untersuchen, wie verschiedene Populationen miteinander verwandt sind, wie sie mit kalt-temperierten Meeren zurechtkommen oder wie menschliche Einflüsse ihre genetische Gesundheit beeinträchtigen könnten.
Ein vollständiger DNA-Bauplan entsteht
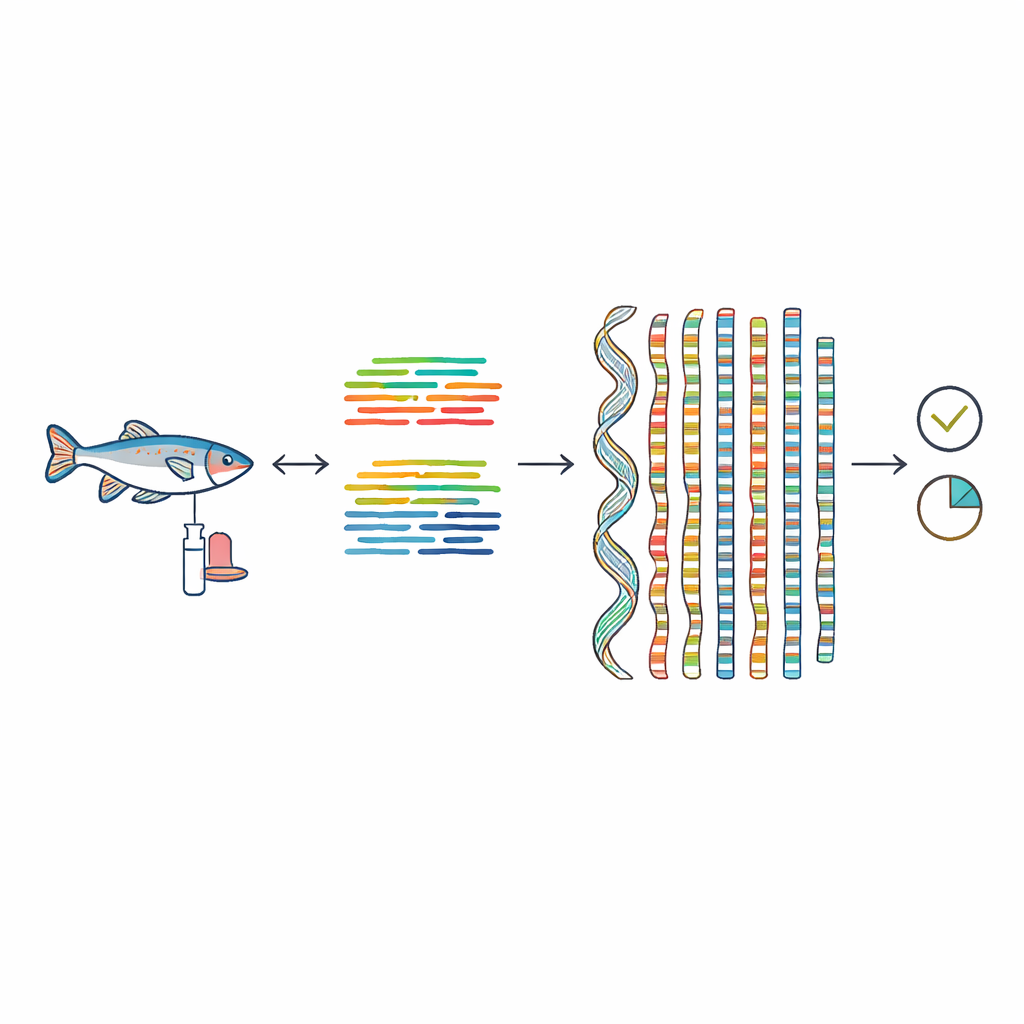
Um diese Lücke zu schließen, sammelten die Forscher einen einzelnen männlichen Fisch von der Küste Qingdaos in China und konservierten sorgfältig mehrere Gewebeproben. Aus seiner Muskulatur isolierten sie lange, intakte DNA-Moleküle und sequenzierten diese mit einem PacBio-HiFi-Sequenzer, der sehr lange DNA-Abschnitte mit hoher Genauigkeit liest. Ergänzend nutzten sie große Mengen kürzerer DNA-Lesesätze von einem Illumina-Sequenzer sowie spezielle Hi-C-Daten, die erfassen, welche DNA-Stücke im Zellkern physisch nahe beieinander liegen. Zusammen ermöglichten diese unterschiedlichen Datenströme, das Genom wie ein hochdetailliertes Puzzle zusammenzusetzen.
Von Fragmenten zu Chromosomen
Mithilfe moderner Assemblersoftware fügte das Team zunächst die langen DNA-Lesesätze zu großen, kontinuierlichen Abschnitten zusammen und entfernte anschließend durch Tiefendeckungs- und Alignment-Schritte redundante Fragmente. Die Hi-C-Daten fungierten dann als eine Art 3D-Karte, die zeigte, welche DNA-Abschnitte auf demselben Chromosom liegen und in welcher Reihenfolge. Mit zusätzlichen manuellen Kontrollen ergab dieser Prozess ein Genom von etwa 618 Millionen DNA-Basen, von dem nahezu der gesamte Anteil (98,51 %) auf 28 Chromosomen zugeordnet werden konnte. Viele dieser Chromosomen reichen bis zu einem oder beiden natürlichen Enden, den Telomeren, was darauf hindeutet, dass die Assembly sehr nahe an die tatsächlichen physischen Grenzen der Chromosomen heranreicht.
Gene, Wiederholungen und Genomqualität
Sobald die Grundstruktur stand, identifizierten die Wissenschaftler, was die DNA tatsächlich kodiert. Zuerst maskierten sie repetitive Elemente — DNA-Abschnitte, die vielfach im Genom vorkommen und nahezu 39 % seiner Länge ausmachen, dominiert von DNA-Transposons und anderen mobilen Elementen. Auf der bereinigten Sequenz kombinierten sie drei Evidenzquellen zur Genprognose: Computermodelle, Vergleiche mit bekannten Proteinen verwandter Fische und RNA-Sequenzen aus fünf Geweben desselben Individuums. Dieser mehrgleisige Ansatz lieferte 22.165 proteinkodierende Gene, von denen mehr als 98 % in bekannten Protein- oder Funktionsdatenbanken zugeordnet werden konnten. Außerdem katalogisierten sie Tausende nicht-kodierender RNA-Gene, wie microRNAs und Transfer-RNAs, die bei der Regulation und dem grundlegenden zellulären Betrieb helfen.
Das Genom auf dem Prüfstand
Um sicherzustellen, dass dieser neue Bauplan verlässlich ist, führte das Team eine Reihe von Qualitätsprüfungen durch. Sie untersuchten, wie häufig in Strahlenflossern erwartete Standard-Referenzgene in der Assembly vorkamen und fanden, dass mehr als 98 % vorhanden und nahezu alle vollständig waren. Unabhängige Werkzeuge zur Schätzung von Fehlerraten gaben dem Genom eine hohe Konsensus-Qualitätsbewertung, und sowohl lange als auch kurze DNA-Lesesätze konnten nahezu perfekt auf die Assembly gemappt werden. Hi-C-Kontaktkarten zeigten starke, saubere Chromosomenmuster und bestätigten damit weiter, dass die großskalige Struktur solide ist.
Bedeutung für Küsten und Naturschutz
Für Nichtfachleute ist die zentrale Erkenntnis, dass Forscher eine hochdetaillierte, chromosomenebene Karte der DNA von Chirolophis japonicus erstellt haben. Diese Ressource macht aus einem einst wenig beachteten Riff-Fisch ein genetisches Modell, um zu untersuchen, wie Küstenarten sich an kalte, sich verändernde Meere anpassen und wie menschliche Aktivitäten ihr langfristiges Überleben beeinflussen. Mit diesem nun öffentlich verfügbaren Genom können Forscher Populationsstrukturen untersuchen, Gene identifizieren, die mit Temperaturtoleranz oder Fortpflanzung verknüpft sind, und bessere Strategien zur Verwaltung und Erhaltung dieses markanten Bewohners nördlicher Felsküsten entwickeln.
Zitation: Liu, K., Liu, Q., Qu, Y. et al. Chromosome-level genome assembly of Chirolophis japonicus Herzenstein, 1890 (Stichaeidae, Perciformes). Sci Data 13, 577 (2026). https://doi.org/10.1038/s41597-026-06893-1
Schlüsselwörter: Meeresgenomik, kalt-temperater Fisch, Chromosomenebene-Assembly, Felsriff-Ökosysteme, Erhaltungsgenetik