Clear Sky Science · de
Aufdeckung des thermodynamisch-kinetischen Trade-off-Effekts an Säurestellen bei zeolithkatalysierter Alkoholdeshydratisierung
Wie Pflanzenalkohol in nützliche Gase verwandelt wird
Viele Pläne für eine sauberere chemische Industrie setzen darauf, Alltagsprodukte aus erneuerbaren Alkoholen wie Ethanol statt aus Rohöl herzustellen. Ein vielversprechender Weg ist die Umwandlung von Ethanol in Ethylen, einen grundlegenden Baustein für Kunststoffe und andere Materialien. Zeolithminerale sind bereits leistungsfähige Katalysatoren für diese Umwandlung, doch Wissenschaftler tun sich weiterhin schwer damit, ihr Wirken exakt zu steuern. Diese Studie blickt auf atomarer Ebene in Zeolithe und zeigt, warum zwei verschiedene Arten von inneren „Hot Spots“ die Reaktion jeweils auf unterschiedliche Weise begünstigen und behindern.
Warum winzige Mineralhohlräume wichtig sind
Alkohole wie Ethanol können aus Kohle, Erdgas oder Biomasse gewonnen werden, und ihre Umwandlung in Ethylen könnte die Abhängigkeit von Erdöl verringern. Zeolithe sind poröse Kristalle, deren innere Kanäle als winzige chemische Fabriken fungieren. In diesen Kanälen sitzen spezielle Säurestellen, die bei Reaktionen die Hauptarbeit leisten. Ein Typ, die Brønsted-Stellen, verhält sich ähnlich wie klassische Protonenlieferanten. Der andere Typ, die Lewis-Stellen, agiert eher wie elektronensuchende Metallzentren. In realen industriellen Katalysatoren koexistieren diese beiden Arten von Stellen meist, was es schwierig macht zu unterscheiden, welche Stelle was bewirkt und wie man sie für sauberere, selektivere Chemie abstimmen kann.
Zwei Arten von Hot Spots, zwei Arten von Hilfe
Die Forschenden bereiteten eine Reihe von ZSM-5-Zeolithen vor, bei denen sie das Verhältnis von Brønsted-reichen, Lewis-reichen und gemischten Materialien einstellen konnten. Mithilfe fortgeschrittener Festkörper-NMR und anderer Spektroskopien beobachteten sie direkt kurzlebige Oberflächenspezies, die entstehen, wenn Ethanol auf diese Säurestellen trifft. An Brønsted-Stellen bildet Ethanol „oberflächengebundene Ethoxy-Spezies“, die dann Wasserstoff abgeben, um Ethylen zu bilden. An Lewis-Stellen bindet Ethanol etwas anders als „chemisorbiertes Ethanol“. Beide Reaktionswege umfassen zwei Hauptetappen: zunächst das Aufbrechen der OH-Bindung des Alkohols zur Bildung einer aktivierten Oberflächenspezies, und anschließend das Entfernen eines Wasserstoffs aus dem Kohlenstoffgerüst zur Freisetzung von Ethylen.
Ein eingebauter Zielkonflikt zwischen Leichtigkeit und Geschwindigkeit
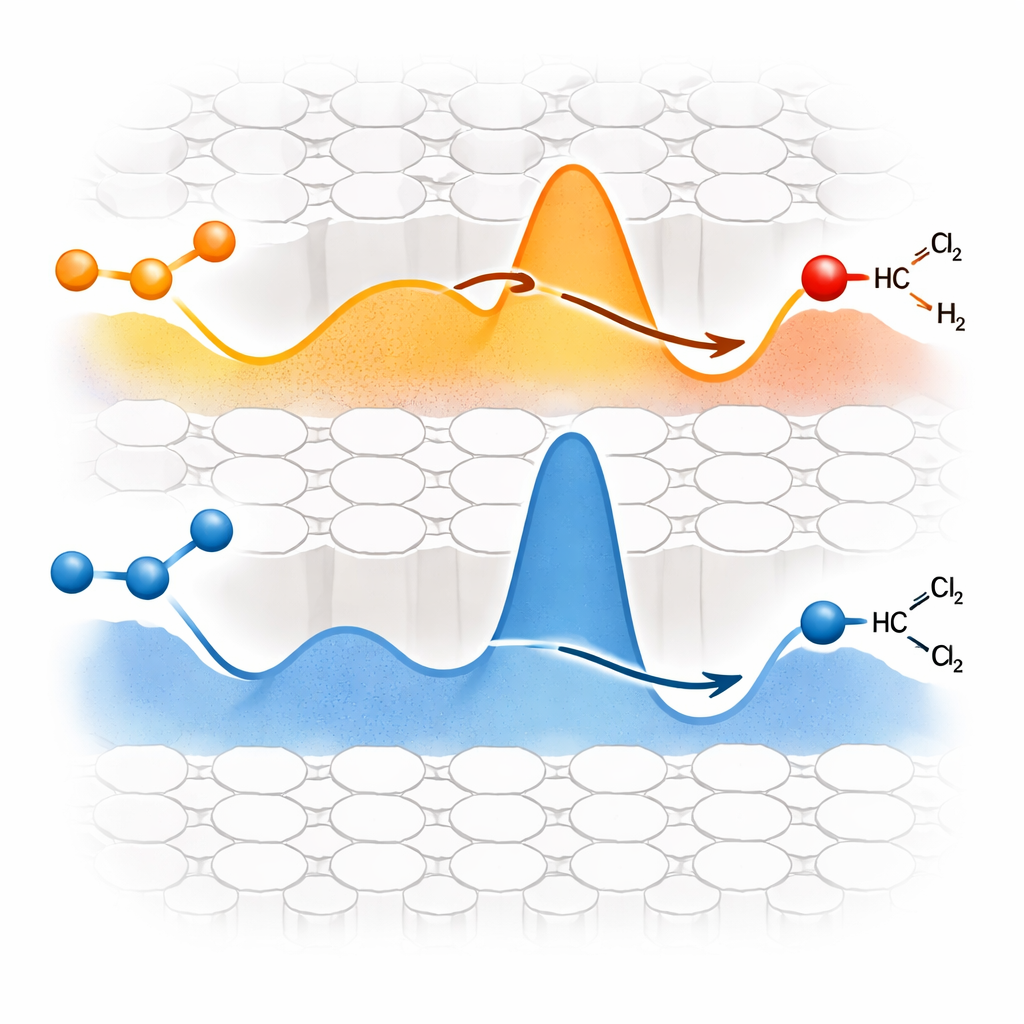
Indem das Team diese Spezies bei steigender Temperatur verfolgte, entdeckte es einen thermodynamisch–kinetischen Zielkonflikt zwischen den beiden Stelltypen. Lewis-Stellen binden Ethanol bereits bei Raumtemperatur leicht und stabilisieren das chemisorbierte Ethanol sehr stark. Das macht den ersten Schritt – die Aktivierung der OH-Gruppe – energetisch günstig. Weil das gebundene Zwischenprodukt jedoch so stabil ist, erfordert der zweite Schritt, in dem es Wasserstoff abgeben muss, um Ethylen freizusetzen, einen großen energetischen Schub und tritt nur bei höheren Temperaturen und langsamer auf. Brønsted-Stellen verhalten sich genau umgekehrt. Sie benötigen mehr Wärme, um zunächst Ethoxy-Spezies zu bilden, doch einmal gebildet, wandeln sich diese Zwischenprodukte relativ leicht in Ethylen um, mit einer niedrigeren Energiebarriere für den zweiten Schritt. Dieses „einfacher erster Schritt, schwerer zweiter Schritt“ versus „schwerer erster Schritt, einfacher zweiter Schritt“-Gegensatz bildet den Kern des Trade-offs.
Theorie mit realer Leistung in Einklang bringen
Computersimulationen mit Dichtefunktionaltheorie kartierten die gesamte Energielandschaft beider Wege und passten gut zu den experimentellen Beobachtungen. Sie zeigten, dass die stärkere Bindung von Ethanol an Lewis-Stellen mit höheren Barrieren für den letzten Wasserstoffentfernungs-Schritt einhergeht. Brønsted-Stellen, obwohl anfangs weniger einladend, bieten hingegen einen reibungsloseren Weg zu Ethylen, sobald das Zwischenprodukt gebildet ist. Kinetische Messungen an Brønsted-reichen und Lewis-reichen Zeolithen bestätigten die vorhergesagten Aktivierungsenergien. Interessanterweise zeigt sich dasselbe Muster auch bei der Dehydratisierung von Isopropanol zu Propen, was darauf hindeutet, dass dieser Zielkonflikt ein allgemeines Merkmal der Alkoholdeshydratisierung auf Zeolithen und kein spezielles Phänomen von Ethanol ist.
Schlauere Katalysatoren aus diesem Gleichgewicht entwerfen
Für Laien lautet die Kernbotschaft: Nicht alle „Säurestellen“ innerhalb eines Zeoliths unterstützen eine Reaktion auf dieselbe Weise. Der eine Typ macht es sehr leicht, dass Alkoholmoleküle haften und die Reaktion beginnen, verlangsamt aber die endgültige Freisetzung des nützlichen Produkts. Der andere Typ ist schwerer anzustoßen, erlaubt jedoch ein schnelleres Abschluss der Reaktion, sobald sie läuft. Indem Katalysatordesigner dieses eingebaute Geben und Nehmen erkennen, können sie auf eine sorgfältig ausbalancierte Mischung aus Brønsted- und Lewis-Stellen zielen, die beide Reaktionsstadien optimiert. Diese Einsicht liefert eine Roadmap zur Schaffung effizienterer, selektiverer und langlebigerer Katalysatoren zur Umwandlung erneuerbarer Alkohole in wichtige chemische Bausteine.
Zitation: Hu, M., Chu, Y., Wang, C. et al. Unveiling the thermodynamic-kinetic trade-off effect on acid sites in zeolite-catalyzed alcohol dehydration. Nat Commun 17, 3675 (2026). https://doi.org/10.1038/s41467-026-70418-y
Schlüsselwörter: Zeolithkatalyse, Ethanoldeshydratisierung, Säurestellen, Ethylenproduktion, Festkörper-NMR