Clear Sky Science · de
Lange Read-Sequenzierung von Familien zeigt erhöhte Keimbahn- und postzygotische Mutationsraten in repetitiver DNA
Warum winzige DNA-Änderungen für Familien wichtig sind
Jedes Kind trägt eine Handvoll genetischer Veränderungen, die bei keinem der beiden Elternteile vorkommen. Die meisten sind harmlos, doch einige können Gesundheit und Entwicklung beeinflussen. Jahrelang hatten Forschende Schwierigkeiten, diese frischen Mutationen genau zu erfassen, weil viele von ihnen in den repetitivsten, schwer lesbaren Bereichen unseres Genoms verborgen liegen. Diese Studie nutzt neue Long-Read-Sequenzierungsverfahren in realen Familien, um jene versteckten Änderungen aufzudecken und zu untersuchen, wo und wann im Leben sie entstehen.
DNA mit weiterem Blick lesen
Traditionelle DNA-Sequenzierung zerteilt das Genom in kurze Fragmente, die dann wie ein Puzzle wieder zusammengesetzt werden. Dieser Ansatz funktioniert für den Großteil unserer DNA gut, versagt aber in langen, wiederholten Abschnitten, in denen viele Teile nahezu identisch aussehen. Die Autor:innen kombinierten drei Technologien – zwei Long-Read-Plattformen und übliches Short-Read-Sequencing – um 73 Kinder und ihre Eltern aus 42 Familien zu untersuchen, überwiegend rekrutiert aus Autismusstudien, in denen keine klare genetische Ursache gefunden worden war. Durch den Vergleich jedes Kindesgenoms mit denen beider Eltern und die plattformübergreifende Überprüfung stellten sie einen Katalog hochvertrauter, für jedes Kind einzigartiger de-novo-Mutationen zusammen.
Im untersuchten, zuverlässig analysierbaren Teil des Genoms – etwa 92 Prozent unserer 2,9 Milliarden DNA-Basen – fanden die Forschenden im Mittel 95 neue Mutationen pro Kind. Die meisten waren Einzelbasenveränderungen; ein geringerer Anteil waren kurze Insertionen oder Deletionen. Trotz der Auswahl der Familien über Autismus trugen betroffene Kinder und ihre nicht betroffenen Geschwister ähnliche Anzahl und Typen neuer Mutationen. Das legt nahe, dass in diesen Familien das Autismusrisiko eher von der Position spezifischer seltener Mutationen abhängt als von einer allgemeinen Erhöhung der Mutationslast.
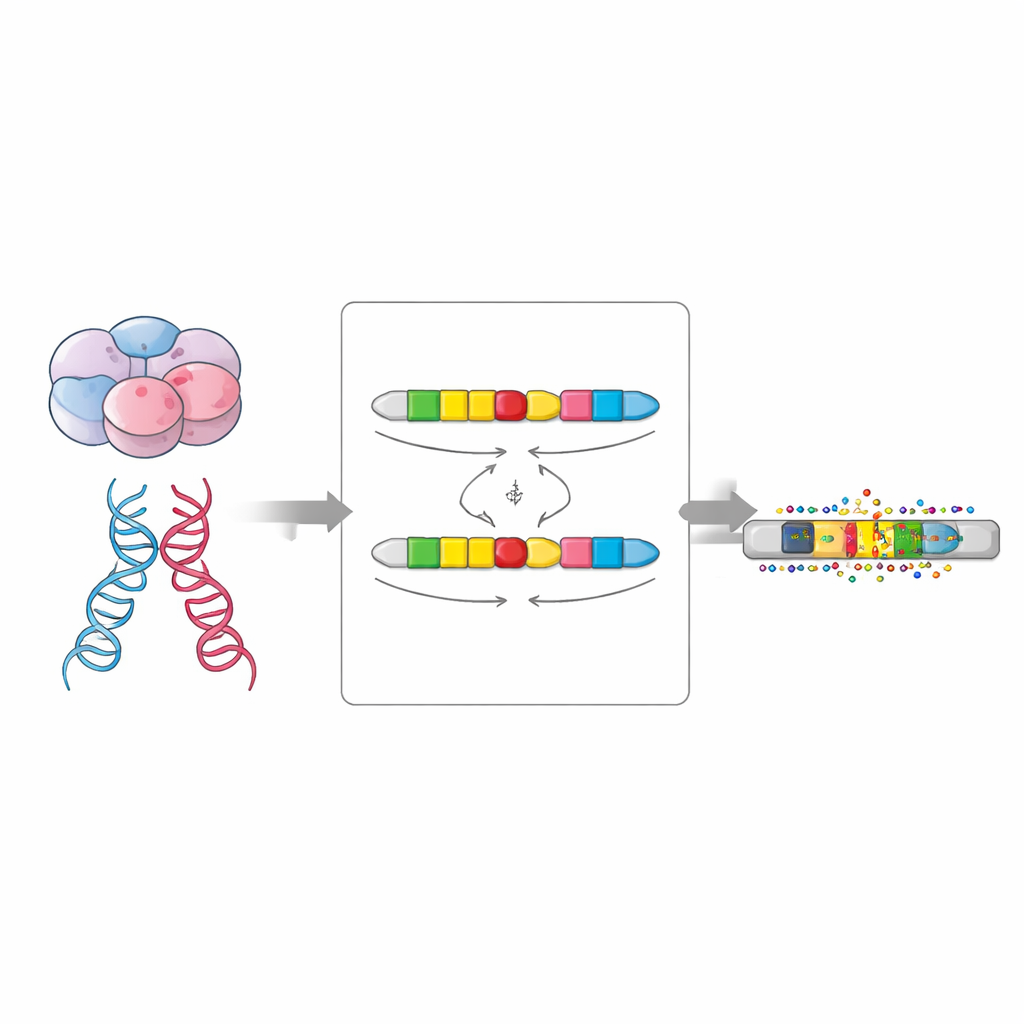
Wann Mutationen entstehen: vor oder nach der Befruchtung
Neue Mutationen können in zwei groben Zeitfenstern auftreten. Manche entstehen in den Eizellen und Spermien der Eltern und liegen damit in jeder Zelle des Kindes vor; das sind Keimbahnmutationen. Andere tauchen kurz nach der Befruchtung in den ersten Zellteilungen auf, sodass nur ein Teil der Körperzellen sie trägt; das sind postzygotische bzw. frühembryonale Mutationen. Lange DNA-Reads sind groß genug, um viele vererbte Marker gleichzeitig zu überbrücken, wodurch die Forschenden nahezu jede neue Mutation der mütterlichen oder väterlichen Chromosomkopie zuordnen und erkennen konnten, ob sie in allen Kopien oder nur in einer Untergruppe vorkommt – ein entscheidender Hinweis auf ihr Entstehungszeitfenster.
Das Team schätzte eine Keimbahnmutationsrate von etwa 1,3 × 10⁻⁸ Substitutionen pro DNA-Basis und Generation, was mit früheren Arbeiten übereinstimmt, und eine postzygotische Rate von etwa einem Sechstel dieser Größe. Ungefähr 15 Prozent der Einzelbasenänderungen entstanden nach der Befruchtung – fast doppelt so viele wie viele frühere Schätzungen, die nur auf Short-Read-Daten beruhten. Wie in früheren Studien stammten die meisten Keimbahnmutationen vom Vater, und ihre Zahl stieg mit dem Alter beider Elternteile, ausgeprägter beim Vater. Postzygotische Mutationen zeigten nur eine leichte väterliche Schieflage und einen schwächeren Alterseffekt, was auf unterschiedliche biologische Prozesse im frühen Embryo hindeutet.
Repetitive DNA als Brennpunkt für Veränderungen
Ein zentrales Ziel der Studie war die Frage, ob repetitive DNA – etwa mobile Elemente und lange duplizierte Segmente – schneller mutiert als der Rest des Genoms. Long-Read-Daten erlauben es nun, diese Regionen direkt zu untersuchen, anstatt sie auszuschließen. Die Autor:innen fanden, dass bestimmte Repeats, namentlich SINE-Elemente wie Alus und große duplizierte Blöcke, sogenannte segmentale Duplikationen, deutlich erhöhte Mutationsraten aufweisen. Innerhalb dieser Duplikationen gilt: je ähnlicher und länger die Kopien, desto höher die Mutationsrate, insbesondere für Veränderungen, die nach der Befruchtung auftreten.
Postzygotische Mutationen waren in hochähnlichen Duplikationen und in den repetitiven Kernen der Chromosomen, den Zentromeren, mehr als doppelt so häufig wie in gewöhnlicher DNA. Das Muster der Basenveränderungen in diesen Hotspots unterschied sich von typischen Keimbahnmutationen: Es gab weniger der klassischen altersbedingten CpG-Veränderungen und mehr Transversionen, also Wechsel zwischen den chemischen Basenklassen. Die Autor:innen argumentieren, dass fehlerhafte DNA-Reparatur und ein Prozess namens Genkonversion – bei dem eine wiederholte Kopie eine andere überschreibt – diese Überschuss an Mutationen in Repeats während der frühesten Entwicklungsstadien antreiben könnten.
Was das für unser Verständnis von Mutationen bedeutet
Indem sie Long-Read-Sequenzierung in realen Familien nutzten, zeigt diese Arbeit, dass unser Genom in repetitiver DNA mehr neue Mutationen ansammelt als bislang angenommen und dass viele dieser Veränderungen kurz nach der Befruchtung entstehen – nicht nur in den Eizellen und Spermien der Eltern. Die Gesamtrate der Genomveränderung pro Generation fällt moderat höher aus, sobald diese frühembryonalen Mutationen berücksichtigt werden; klassische Short-Read-Ansätze haben offenbar einen beträchtlichen Anteil übersehen – besonders in komplexen Repeats. Für Nicht‑Fachleute lautet die Kernbotschaft: Die lange als zu repetitiv zum Studieren abgestempelte „Dunkle Materie“ des Genoms ist aktiver und bedeutsamer für Mutationen, als wir dachten. Zu verstehen, wie sich diese Regionen im Lauf der Zeit verändern, wird entscheidend sein, um genetische Variation und ihre Verknüpfung mit Krankheiten zu deuten.
Zitation: Noyes, M.D., Sui, Y., Kwon, Y. et al. Long-read sequencing of families reveals increased germline and postzygotic mutation rates in repetitive DNA. Nat Commun 17, 3717 (2026). https://doi.org/10.1038/s41467-026-70342-1
Schlüsselwörter: de-novo-Mutationen, Long-Read-Sequenzierung, repetitive DNA, segmentale Duplikationen, postzygotische Mosaike