Clear Sky Science · de
NEDAMSS-Syndrom‑assoziierte truncierende und Missense‑Mutationen sind mit aberranter Flüssig‑Flüssig‑Phasentrennung von IRF2BPL verbunden
Wenn Proteine aus dem Gleichgewicht geraten
NEDAMSS ist eine seltene Kindererkrankung, bei der betroffene Kinder Fähigkeiten verlieren, die sie zuvor besaßen, und die zu Bewegungsstörungen, Sprachverlust und Anfällen führt. Bis vor Kurzem wussten Ärztinnen und Ärzte zwar, dass die Erkrankung mit Veränderungen in einem wenig verstandenen Gen namens IRF2BPL verknüpft ist, nicht aber, wie diese Veränderungen die Nervenzellen schädigen. Diese Studie zeigt, dass der Übeltäter nicht einfach ein fehlfunktionelles Protein ist, sondern ein Protein, das sich innerhalb von Nervenzellen zunehmend wie winzige Flüssigkeitströpfchen verhält und dadurch letztlich die Funktion und das Überleben von Neuronen stört. 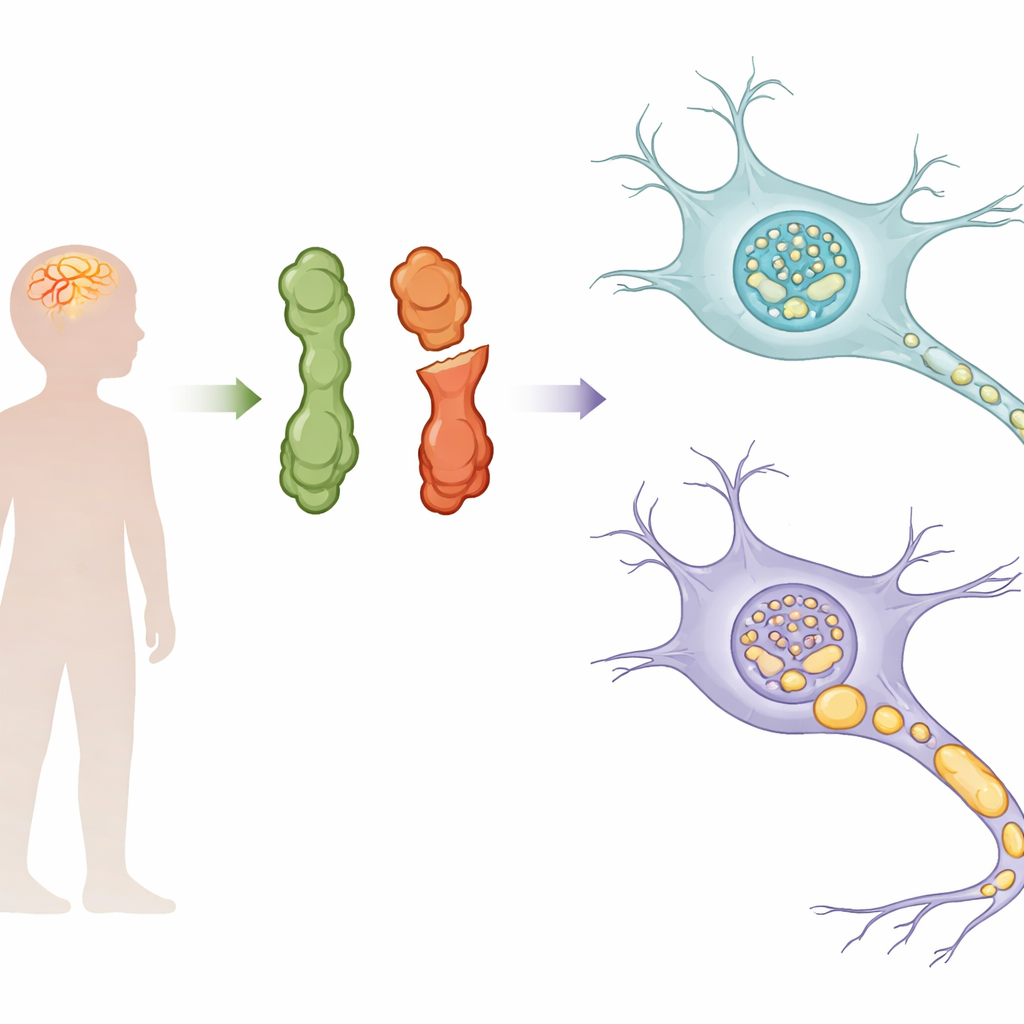
Ein wenig bekanntes Gen mit großer Bedeutung
Das IRF2BPL‑Gen kodiert ein Protein, das bei der Steuerung aktiviert‑/deaktivierter Gene mitwirkt, besonders im Gehirn. Jahrelang galt es in Datenbanken als „wenig untersucht“ mit unklarer Funktion. Die meisten Patientinnen und Patienten mit NEDAMSS tragen Veränderungen, die das Protein verkürzen (truncierende Mutationen) oder eine Aminosäure gegen eine andere austauschen (Missense‑Mutationen). Auffällig ist, dass diese Veränderungen in der langen mittleren Region des Proteins gehäuft auftreten, einem Bereich, der reich an einfachen Aminosäurenwiederholungen ist und früher als strukturarm („low‑complexity“) abgetan wurde. Die Autorinnen und Autoren zeigen, dass diese zentrale Region tatsächlich in drei einfache Segmente und eine strukturiertere Domäne unterteilt ist und dass diese Organisationsstruktur über Hunderte von Wirbeltierarten hinweg konserviert ist, was auf wichtige biologische Funktionen hinweist.
Proteine, die sich wie winzige Flüssigkeiten verhalten
In Zellen sitzen manche Proteine nicht isoliert oder in Membranen, sondern sammeln sich zu tropfenähnlichen Kondensaten durch einen Prozess namens Flüssig–Flüssig‑Phasentrennung, vergleichbar mit Öltröpfchen in Wasser. Die Forschenden fanden heraus, dass IRF2BPL normalerweise solche Tröpfchen in vielen Zelltypen bildet, einschließlich humaner Neuronen, die aus Stammzellen gezüchtet wurden. Mithilfe hochauflösender Mikroskopie beobachteten sie kleine, runde Kondensate sowohl im Zellkern, wo die DNA liegt, als auch entlang von Axonen und Synapsen. Diese Tröpfchen reagierten empfindlich auf Chemikalien, die schwache Wechselwirkungen stören, erholten sich schnell nach Photobleaching und konnten innerhalb von Minuten verschmelzen und sich wieder auflösen — alles klassische Merkmale flüssigkeitsähnlichen Verhaltens statt starrer Proteinaggregate. Ein Zink‑Finger‑Segment an einem Ende des Proteins sowie eine benachbarte Alanin‑/Glutamin‑reiche Sequenz erwiesen sich als treibende Einheit dieser Tröpfchenbildung. 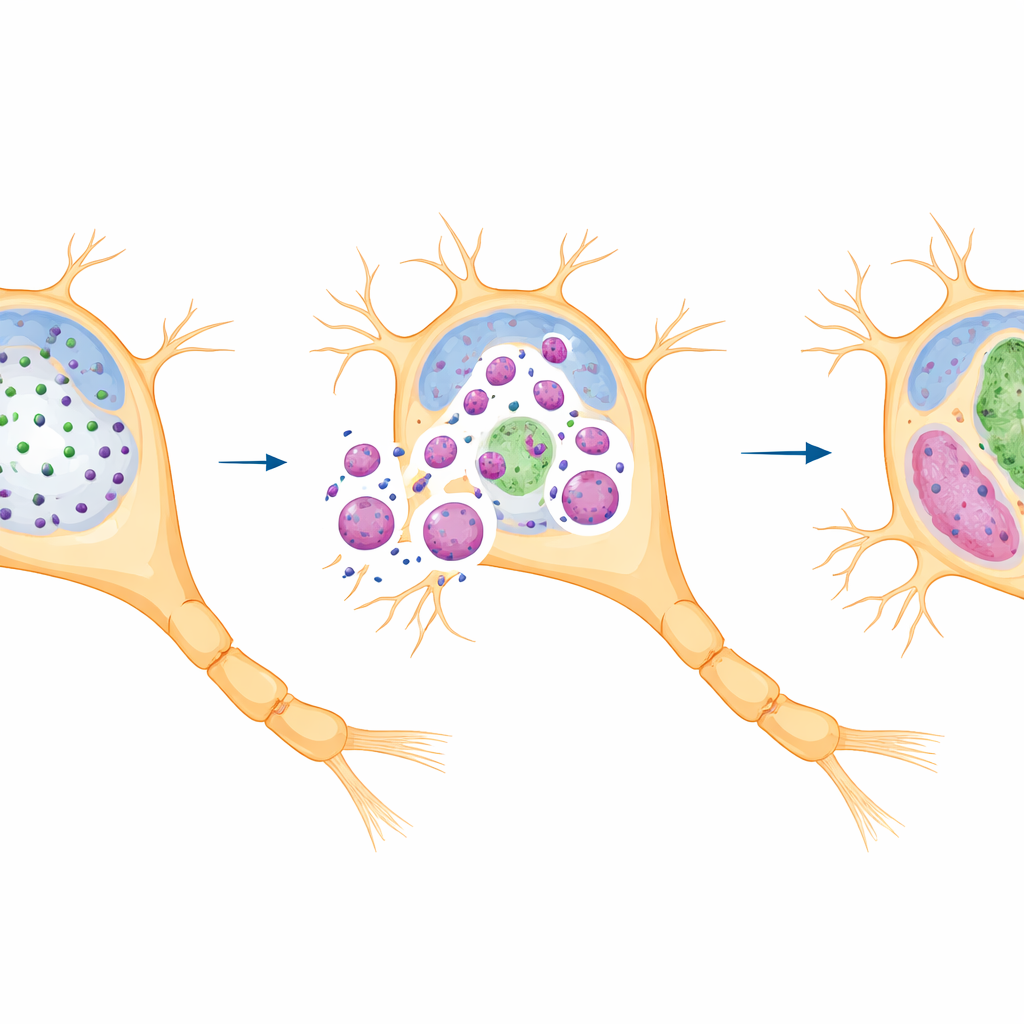
Von gesunden Tröpfchen zu schädlichen Klumpen
Patientenartige Mutationen veränderten diese Tropfenlandschaft massiv. Truncierende Mutationen, die das Protein innerhalb seiner zentralen Wiederholungsregion zerschnitten, erzeugten verkürzte Fragmente, die zwar noch Kondensate bildeten, jedoch mit sehr anderen Eigenschaften. Anstelle vieler kleiner, dynamischer Tröpfchen sammelten sich in den Zellen weniger, größere und häufig längliche Strukturen, die überwiegend im Zytoplasma statt im Zellkern lagen. Diese mutanten Kondensate fusionierten schneller, ließen sich schlechter auflösen und tauschten Moleküle mit ihrer Umgebung langsamer aus, was auf eine Verschiebung von einem flüssigen zu einem gel‑ähnlichen oder fibrillären Zustand hindeutet. Unter dem Elektronenmikroskop enthielten diese Tröpfchen geordnete innere Fasern anstelle des amorphen Inneren normaler Kondensate, was auf eine gefährliche Progression von reversiblen Tröpfchen zu beständigeren, festeren Assemblierungen schließen lässt.
Das gesunde Protein an den falschen Ort ziehen
Eine entscheidende Entdeckung war, dass diese abnormen Kondensate wie molekulare Fallen wirken. Die mutanten IRF2BPL‑Fragmente — insbesondere die kürzeren — zogen das normale Protein, das von der gesunden Genkopie stammte, an und hielten es im Zytoplasma fest. Infolgedessen wurden Kern und Axone der Zellen von funktionsfähigem IRF2BPL entleert. Missense‑Mutationen in der strukturierten mittleren Domäne oder im Zink‑Finger zeigten ein ähnliches Muster: Sie veränderten zwar nicht die Gesamtmenge an Kondensaten, verschoben die Tröpfchen aber aus dem Zellkern ins Zytoplasma und verringerten dadurch erneut den nukleären Pool. Die Fähigkeit der Mutanten, das normale Protein zu rekrutieren, nahm zu, je stärker die Schlüsselwiederholungsregion verkürzt war, was auf einen längenabhängigen Gewinn schädlichen Verhaltens statt auf einen einfachen Funktionsverlust hindeutet.
Von fehlplatzierten Tröpfchen zu fehlregulierten Genen und Neuronen
Als das Team menschliche Zellen so veränderte, dass sie eine krankheitsähnliche Trunkierung in einer IRF2BPL‑Kopie trugen, beobachteten sie, dass das verbleibende normale Protein in zytoplasmatische Kondensate hineingezogen wurde und ein bekanntes Zielgen, WNT1, ungewöhnlich aktiviert war. Bemerkenswerterweise trat derselbe Anstieg von WNT1 auf, wenn IRF2BPL vollständig ausgeschaltet wurde, was zeigt, dass die Sequestrierung des normalen Proteins einem vollständigen Funktionsverlust gleichen kann. In neuronähnlichen Zellen veränderte die Expression von mutanten IRF2BPL das elektrische Ruhezustands‑Potential und verringerte die Größe der Nervenimpulse, was auf beeinträchtigte neuronale Erregbarkeit und frühe Schädigungszeichen hinweist. Zusammen verbinden diese Befunde fehlverhaltende Kondensate direkt mit Genfehlregulation und gestörter Neuronfunktion und liefern eine schlüssige Kette vom Mutationsereignis bis zur Zelltörung.
Warum das für Kinder mit NEDAMSS wichtig ist
Für Familien, die mit NEDAMSS und verwandten IRF2BPL‑Erkrankungen konfrontiert sind, bietet diese Arbeit eine vereinheitlichende Erklärung: Die Krankheit entsteht nicht nur durch fehlendes Protein, sondern durch ein Protein, das die falsche Art von Tröpfchen am falschen Ort bildet. Mutationen verschieben IRF2BPL von kleinen, flexiblen nukleären Tröpfchen hin zu großen, stabilen zytoplasmatischen Kondensaten, die das gesunde Protein aufsaugen, seine normale Funktion in der Genregulation lahmlegen und die neuronale Signalübertragung stören. Die Erkenntnis, dass aberrante Phasentrennung einen zentralen Mechanismus darstellt, eröffnet neue therapeutische Wege, etwa Substanzen, die das Verhalten von Kondensaten verändern, mutant‑normale Interaktionen verhindern oder die korrekte nukleäre Lokalisation von IRF2BPL wiederherstellen — mit dem langfristigen Ziel, die Gehirnfunktion betroffener Kinder zu erhalten.
Zitation: Dell’Oca, M., Boggio Bozzo, S., Vaglietti, S. et al. NEDAMSS syndrome-related truncating and missense mutations are associated with aberrant liquid-liquid phase separation of IRF2BPL. Nat Commun 17, 3301 (2026). https://doi.org/10.1038/s41467-026-69781-7
Schlüsselwörter: NEDAMSS‑Syndrom, IRF2BPL, Flüssig–Flüssig‑Phasentrennung, Proteinkondensate, Neuroentwicklungsstörungen