Clear Sky Science · de
Proteasom‑Dysfunktion liegt der HERC2-assoziierten neuroentwicklungsstörung mit Angelman‑ähnlichen Merkmalen zugrunde
Wenn das zelluläre Aufräumteam versagt
Unsere Nervenzellen sind auf ein fein abgestimmtes System angewiesen, das kontinuierlich Proteine aufbaut und abbaut. Diese Studie untersucht, was passiert, wenn ein Teil dieses zellulären Aufräumteams durch Veränderungen in einem Gen namens HERC2 gestört ist. Solche Veränderungen finden sich bei Kindern mit einer seltenen Erkrankung, die dem Angelman‑Syndrom sehr ähnelt, mit Entwicklungsverzögerungen, Bewegungsstörungen und autistischen Zügen. Indem die Forscher beobachteten, wie Zellen mit Tausenden verschiedener Proteine umgehen, entdeckten sie, wie Fehler in HERC2 die Hauptrecycling‑Maschine der Zelle, das Proteasom, stören und wie dies zur Erkrankung beitragen kann.
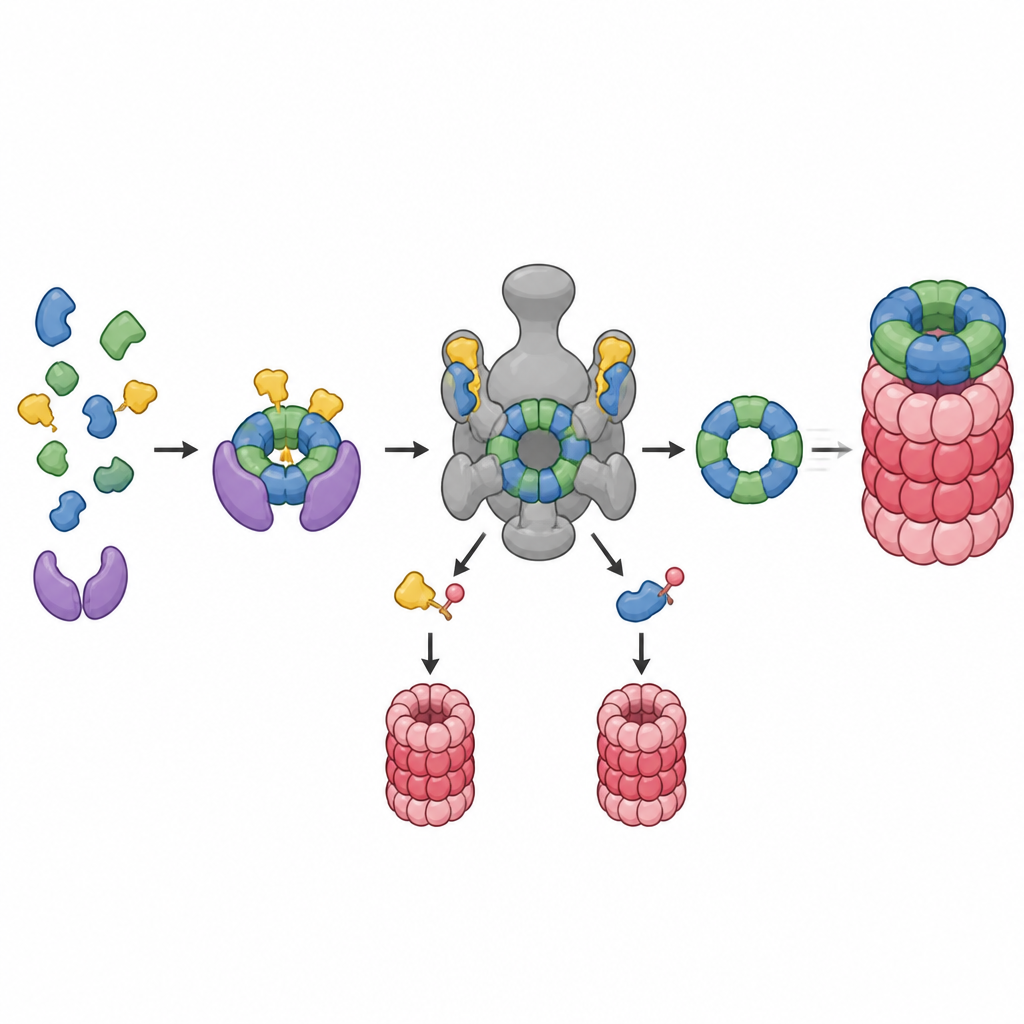
Wie Zellen Proteine normalerweise markieren und recyceln
Im Inneren unserer Zellen werden abgenutzte oder fehlgefaltete Proteine mit winzigen molekularen Markern versehen und anschließend in das Proteasom eingespeist, einen fassförmigen Komplex, der sie in Stücke zerlegt. HERC2 ist eines der Enzyme, die diese Marker anbringen. Um herauszufinden, welche Proteine von HERC2 abhängen, verwendete das Team einen eleganten Trick: Sie versorgten Zellen mit einer biotinmarkierten Version des Markers, sodass alle markierten Proteine herausgefischt und mittels Massenspektrometrie identifiziert werden konnten. Sie verglichen Zellen mit normalem HERC2 mit Zellen, die eine deaktivierte Version ohne Markierungsaktivität produzierten. Dieser direkte Vergleich zeigte, welche Proteine ihre Markierung speziell aufgrund von HERC2 gewannen oder verloren.
Eine verborgene Rolle beim Bau des Proteinzerkleinerers
Der Katalog der markierten Proteine war umfangreich, doch eine Gruppe stach heraus: viele Komponenten des Proteasoms selbst, insbesondere Teile des 19S‑Regulatorischen Partikels, das die Proteine erkennt, entfaltet und in den Kern hineinführt. Die Forscher fanden, dass HERC2 hilft, mindestens elf Proteine zu markieren, die am Aufbau dieser Regulierungs‑Einheit beteiligt sind, darunter mehrere Base‑ und Lid‑Untereinheiten sowie deren Helfer‑Chaperone. Folgeexperimente konzentrierten sich auf eine Untereinheit, PSMC5, und ihren Assemblierungspartner PAAF1. HERC2 bindet an PSMC5 über PAAF1 und markiert nur die unmontierten, überzähligen Kopien zur Zerstörung. Auf diese Weise fungiert HERC2 wie ein Bauinspektor, der dafür sorgt, dass lose Teile, die nicht in die fertige Maschine passen, entfernt und nicht abgelagert werden.
Die Balance der Aktivität des zellulären Zerkleinerers
Wurden HERC2‑Spiegel in Zelllinien im Labor reduziert, sank die Gesamtaktivität des Proteasoms, obwohl seine Grundstruktur noch nachweisbar war. Das deutet darauf hin, dass weniger vollständig montierte Maschinen verfügbar waren. Mit fluoreszenten Reportern zeigte das Team, dass das Entfernen von HERC2 die unmontierte PSMC5‑Untereinheit stabiler macht, was mit dem Versagen übereinstimmt, ungenutzte Teile zu beseitigen. In Hautzellen von Patientinnen und Patienten, die eine häufige HERC2‑Variante tragen, die mit der neuroentwicklungsstörung assoziiert ist, sah die Lage jedoch anders aus: Diese Zellen zeigten eine erhöhte Proteasomaktivität und höhere PSMC5‑Spiegel. Das mutierte HERC2‑Protein war weniger stabil und interagierte schlecht mit dem PSMC5–PAAF1‑Komplex, was darauf hindeutet, dass sowohl der Verlust des Inspektors als auch seine fehlerhafte Bindung an Untereinheiten das Gleichgewicht des Proteinabbaus stören können.
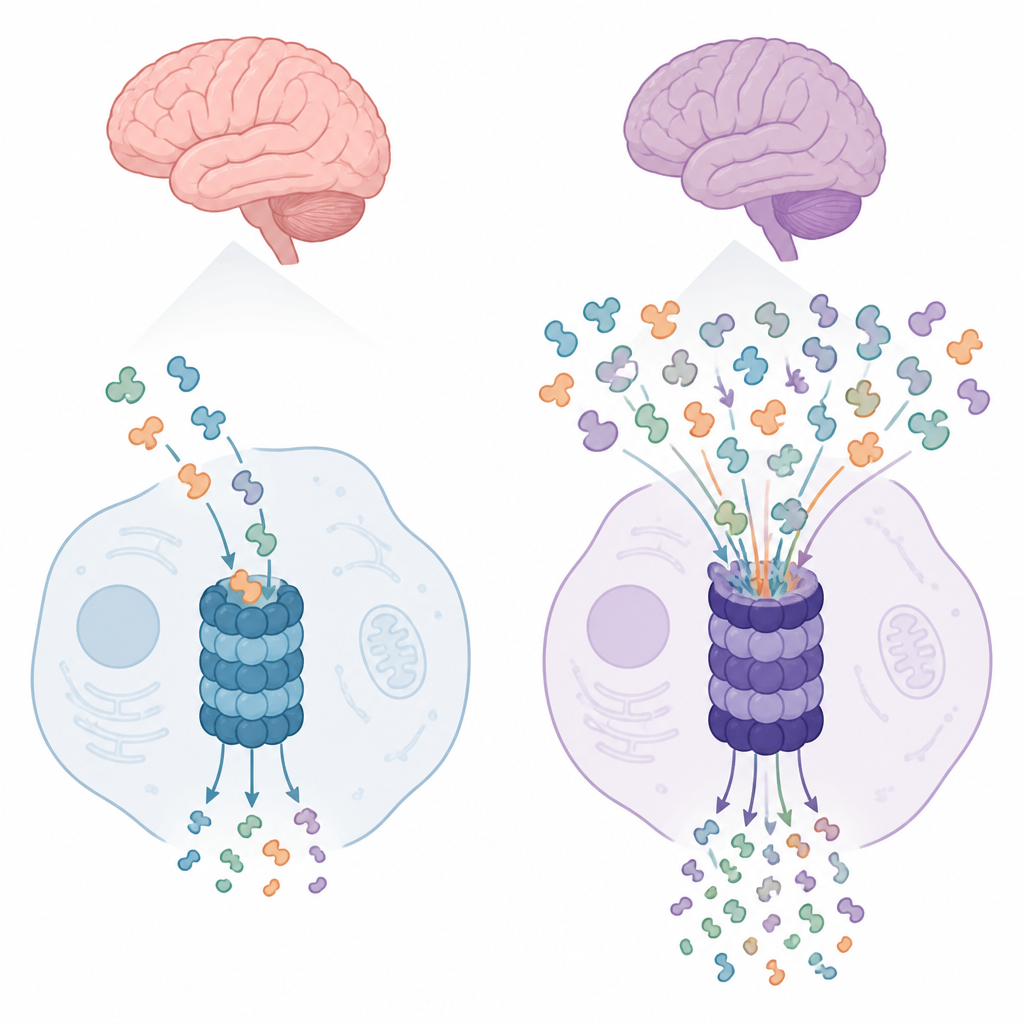
Von zellulärem Ungleichgewicht zu Gehirnsymptomen
Die Befunde fügen sich in ein größeres Bild, in dem HERC‑Familienproteine den Zusammenbau großer Proteinkomplexe in der Zelle steuern – nicht nur des Proteasoms, sondern auch Komplexe, die an Proteinsynthese und Zellstruktur beteiligt sind. Neurone, die ein Leben lang bestehen müssen, sind besonders empfindlich gegenüber langfristigen Ungleichgewichten in der Proteinqualitätskontrolle. Tierstudien haben bereits Defekte verwandter HERC‑Proteine mit dem Absterben bestimmter Nervenzellen und Bewegungsstörungen verknüpft. Hier verbinden die Autorinnen und Autoren eine humane HERC2‑Variante mit verändertem Proteasomverhalten in Patienten‑Zellen und liefern damit eine plausibele Kette vom Gendefekt über gestörtes Proteinrecycling bis hin zu neuroentwicklungsbedingten Symptomen.
Was das für die zukünftige Versorgung bedeutet
Für Nicht‑Spezialisten ist die zentrale Botschaft, dass diese Arbeit einen Schritt der Qualitätskontrolle im zellulären Aufräumsystem identifiziert, der bei einer seltenen Angelman‑ähnlichen Erkrankung fehlreguliert ist. HERC2 entscheidet nicht einfach darüber, ob der Proteinabbau hoch- oder runtergefahren wird, sondern welche unfertigen Teile verworfen werden sollten, damit die Recycling‑Maschine reibungslos läuft. Wenn dieser Entscheidungsprozess versagt, gerät die Proteasomaktivität aus dem Gleichgewicht, was anfällige Nervenzellen belasten kann. Diese Einsichten legen nahe, dass eine präzise Anpassung der Proteasomfunktion oder ein frühzeitiges Screening auf HERC2‑Veränderungen eines Tages helfen könnte, einige Folgen dieser Erkrankung zu behandeln oder zu verhindern – solche Strategien benötigen jedoch noch erheblich mehr Forschung, bevor sie klinisch einsetzbar sind.
Zitation: Sala‑Gaston, J., Costa‑Sastre, L., Garcia‑Diez, M. et al. Proteasome dysfunction underlies HERC2-linked neurodevelopmental disorder with Angelman-like clinical features. Cell Death Discov. 12, 243 (2026). https://doi.org/10.1038/s41420-026-03095-x
Schlüsselwörter: HERC2, Proteasom, neuroentwicklungsstörung, Proteinqualitätskontrolle, Angelman‑ähnliches Syndrom