Clear Sky Science · sv
Hämning av HSP27 aktiverar XBP1s/CerS1‑samspelet, vilket utlöser DRP1‑driven mitofagi och därigenom skyddar mot celldöd och främjar KSHV:s lytiska cykel i celler från primär effusionslymfom
När cellstress blir ett tveeggat svärd
Våra celler överlever dagliga påfrestningar genom att slå på nödlägen som reparerar skador och håller dem vid liv. Cancerceller kan dock kapa dessa samma program för att växa och för att gömma virala följeslagare. Den här artikeln undersöker hur blockering av ett enda skyddsprotein mot stress i ett ovanligt lymfom inte bara knuffar tumörceller mot död utan också ger ett dolt virus inuti dem ett tillfälle att vakna och föröka sig. Att förstå denna känsliga balans kan hjälpa till att utforma behandlingar som dödar cancer samtidigt som de hindrar viruset från att spridas.
Ett dolt virus i ett aggressivt lymfom
Primär effusionslymfom är en starkt aggressiv cancersjukdom i B‑celler, en typ av vita blodkroppar. De flesta av dessa tumörceller bär på en vilande passagerare: Kaposis sarkom‑associerat herpesvirus (KSHV). I sitt tysta, latenta stadium producerar viruset bara några få proteiner och gömmer sig i värdcellens genom. Viss stress kan trycka det in i en aktiv, lytisk fas där det kopierar sig själv och bildar nya virala partiklar, vilket ofta dödar värdcellen. Tumörcellerna själva är beroende av flera stressresponssystem, inklusive så kallade värmechockproteiner och det ofolda proteinsvar (unfolded protein response), som hjälper dem hantera felveckade proteiner, störd lipidmetabolism och skador på sina energiproducerande mitokondrier. 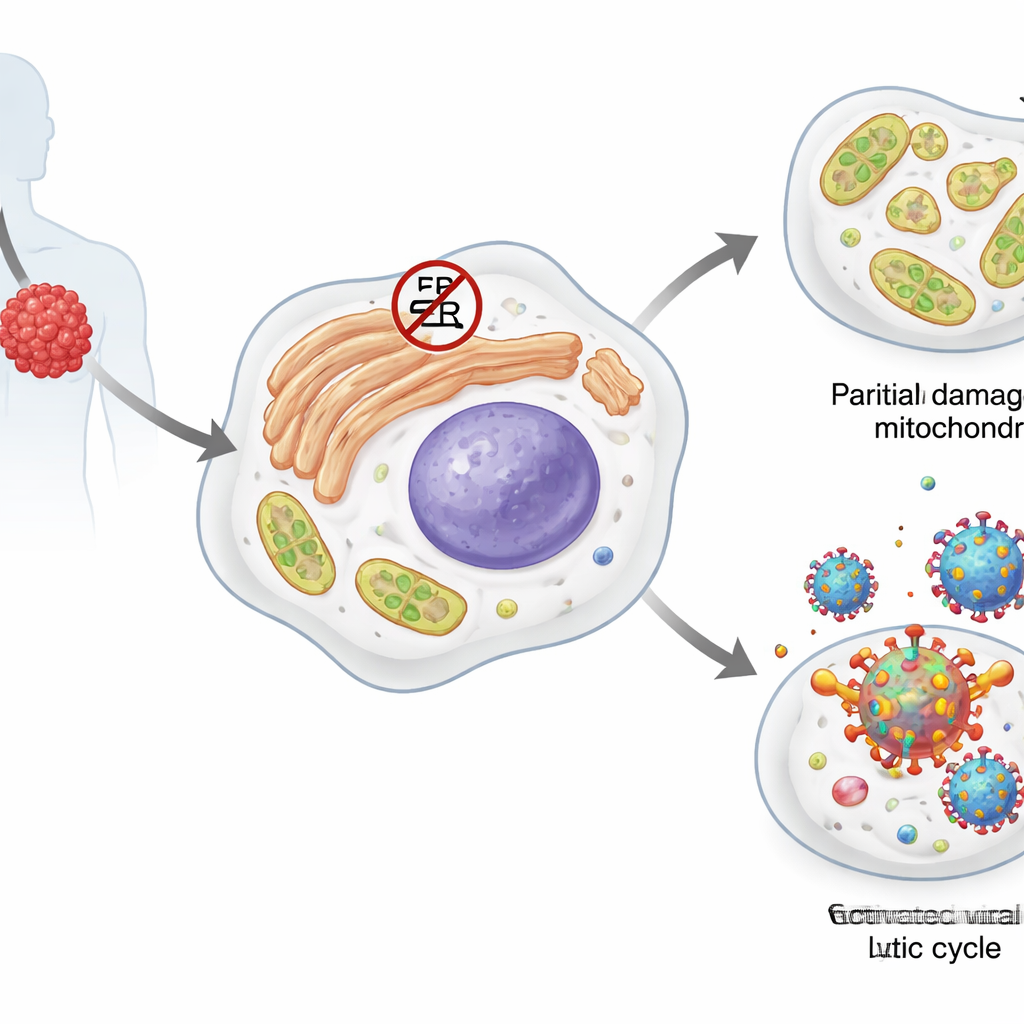
Blockera en cellär livvakt
Forskarna fokuserade på HSP27, ett litet värmechockprotein känt för att skydda celler mot stress. Genom att använda en kemisk hämmare kallad J2 eller genom att tysta genen med små RNA‑molekyler minskade de HSP27‑aktiviteten i lymfomceller odlade i laboratoriet. Detta gjorde cellerna mindre benägna att överleva och utlöste en stark stressignal i ett inre membrannätverk kallat det endoplasmatiska retiklet. Markörer för detta svar, inklusive skyddande och dödsfrämjande faktorer, ökade och en viktig omkopplare kallad XBP1s aktiverades. Samtidigt visade cellerna fler tecken på programmerad celldöd, vilket bekräftar att borttagandet av HSP27 skjuter dem mot en brytpunkt mellan överlevnad och undergång.
En stressloop som kommunicerar med cellfetter
Stress i det endoplasmatiska retiklet är tätt sammanflätad med hur celler hanterar lipider. Teamet fann att hämning av HSP27 ökade nivåerna av CerS1, ett enzym som tillverkar en specifik lipidmolekyl kallad C18‑ceramid. När de kemiskt blockerade XBP1s försvann ökningen av CerS1, vilket visar att XBP1s bidrar till att slå på CerS1‑genen under dessa förhållanden. Slående nog ledde inhibering av CerS1 i sin tur till lägre XBP1s, vilket avslöjar en positiv återkopplingsslinga: varje faktor stöder den andra. Detta molekylära handslag omformar inte bara lipidmetabolismen utan förstärker också cellens förmåga att anpassa sig till endoplasmatiskt retikulum‑stress, även när dödssignaler byggs upp.
Mitokondrier återvinns istället för att förstöras
Stress i en del av cellen spiller ofta över på mitokondrierna, de små kraftverken som genererar energi. Efter att HSP27 blockerats producerade lymfomcellerna mer reaktiva syreföreningar, ett tecken på mitokondriell störning, och ökade nivåer av DRP1, ett protein som delar mitokondrier i mindre fragment. Författarna visade att XBP1s–CerS1‑loopen var ansvarig för att höja DRP1. Detta utlöste i sin tur mitofagi, en kvalitetskontrollprocess där skadade mitokondrier omsluts av membraner och levereras till cellens ”återvinningscentraler” kallade lysosomer. Med hjälp av fluorescerande färgämnen och proteinmarkörer bekräftade de att mitokondrier selektivt togs bort. När de kemiskt eller genetiskt blockerade DRP1 minskade denna mitofagi och cellerna dog lättare, vilket betyder att mitokondriell återvinning faktiskt hjälpte de stressade tumörcellerna att hålla ut. 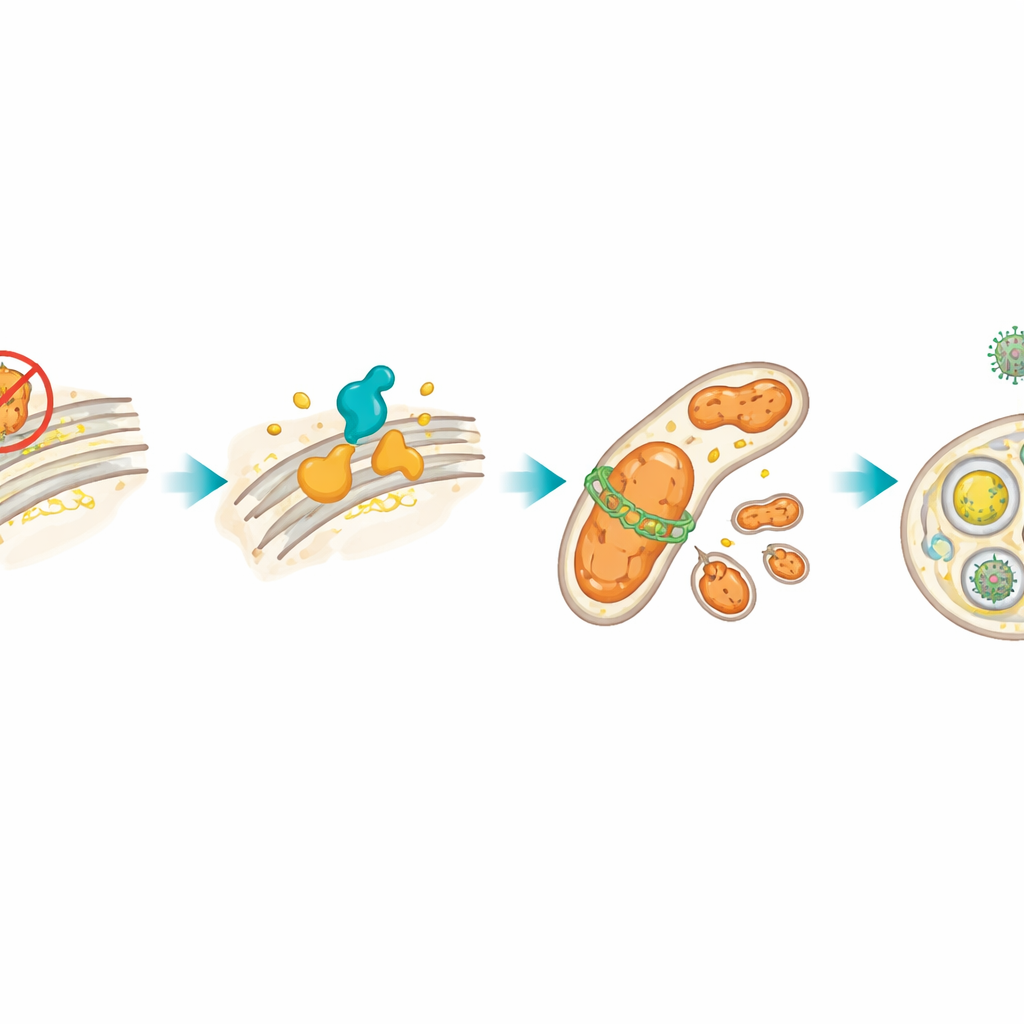
Ger viruset tid att fly
Samma mitofagi som skyddade tumörcellerna gynnade också KSHV. Aktivering av XBP1s, ackumulering av C18‑ceramid och ökad mitokondriell fragmentering har alla kopplats till återuppvaknande av detta virus. Här, när HSP27 hämmades, uttryckte fler celler tidiga och sena virala proteiner, tydliga tecken på lytisk replikation. Blockering av DRP1, och därmed mitofagi, minskade denna virala reaktivering. Författarna föreslår att genom att något förlänga cellöverlevnaden under stress ger mitofagin KSHV tid att slutföra sin replikationscykel och lämna den döende cellen, potentiellt infektera nya målceller och bidra till cancerutveckling.
Vad detta betyder för framtida behandlingar
För en icke‑specialist är huvudbudskapet att HSP27 fungerar som en central trafikledare för hur lymfomceller hanterar stress, hur de återvinner skadade mitokondrier och hur ett cancerkopplat virus bestämmer när det ska vakna. Att stänga av HSP27 släpper lös en kedja av händelser som både underminerar cellöverlevnad och, paradoxalt nog, tillfälligt skyddar cellerna genom mitofagi samtidigt som det tillåter KSHV att replikera. Terapeutiskt kan en kombination av HSP27‑hämning och läkemedel som blockerar DRP1‑driven mitofagi skjuta tumörceller mot snabbare död och begränsa virusets chans att spridas, vilket erbjuder en tvåfaldig strategi mot detta dödliga lymfom.
Citering: Gonnella, R., Corrado, V., Scaffidi, G.F. et al. Inhibiting HSP27 activates the XBP1s/CerS1 interplay, which triggers DRP1-driven mitophagy, thereby protecting against cell death and promoting the KSHV lytic cycle in primary effusion lymphoma cells. Cell Death Discov. 12, 118 (2026). https://doi.org/10.1038/s41420-026-02979-2
Nyckelord: primär effusionslymfom, Kaposis sarkom‑virus, cellstressrespons, mitofagi, värmechockprotein HSP27