Clear Sky Science · sv
En livskraftig kinase-inaktiv RIPK3 D143N-musmodell avslöjar dess stödjande roll i att driva TNF-inducerad inflammatorisk störning
Varför denna musstudie är viktig för förståelsen av inflammation
Många allvarliga sjukdomar, från dödliga infektioner till autoimmuna skov, drivs inte bara av mikrober eller gener utan av organismens egna okontrollerade inflammation. Ett protein kallat RIPK3 har länge setts som en central utförare av en våldsam form av celldöd som eldar på sådan inflammation, vilket gjort det till ett lockande läkemedelsmål. Men RIPK3 har också andra, mindre välkända roller i cellen. Denna studie beskriver en ny laboratoriemus som tydligt skiljer RIPK3:s dödande enzymaktivitet från dess signalgivande "stödjande" roll, visar hur varje funktion bidrar till inflammation och pekar på nya terapeutiska strategier.
Två sätt ett dödsprotein kan verka
Celler kan dö på ordnade eller röriga sätt. Vid ordnad, "tyst" celldöd återvinns cellkomponenter utan större larm. I en rörigare form kallad necroptos spricker celler och läcker ut sitt innehåll, vilket utlöser starka immunsvar. RIPK3 är central för necroptos: när det aktiveras sätter det igång en partner som gör hål i cellmembranet. Tidigare arbete har dock antytt att RIPK3 också kan bidra till mer klassisk, kaspasdriven celldöd och kan förstärka inflammatorisk signalering även utan att döda celler. Att skilja dessa roller åt har varit svårt eftersom befintliga inaktiva varianter antingen dödar embryon eller kraftigt minskar proteinets nivåer, vilket försvårar studier av dess normala scaffold-beteende.
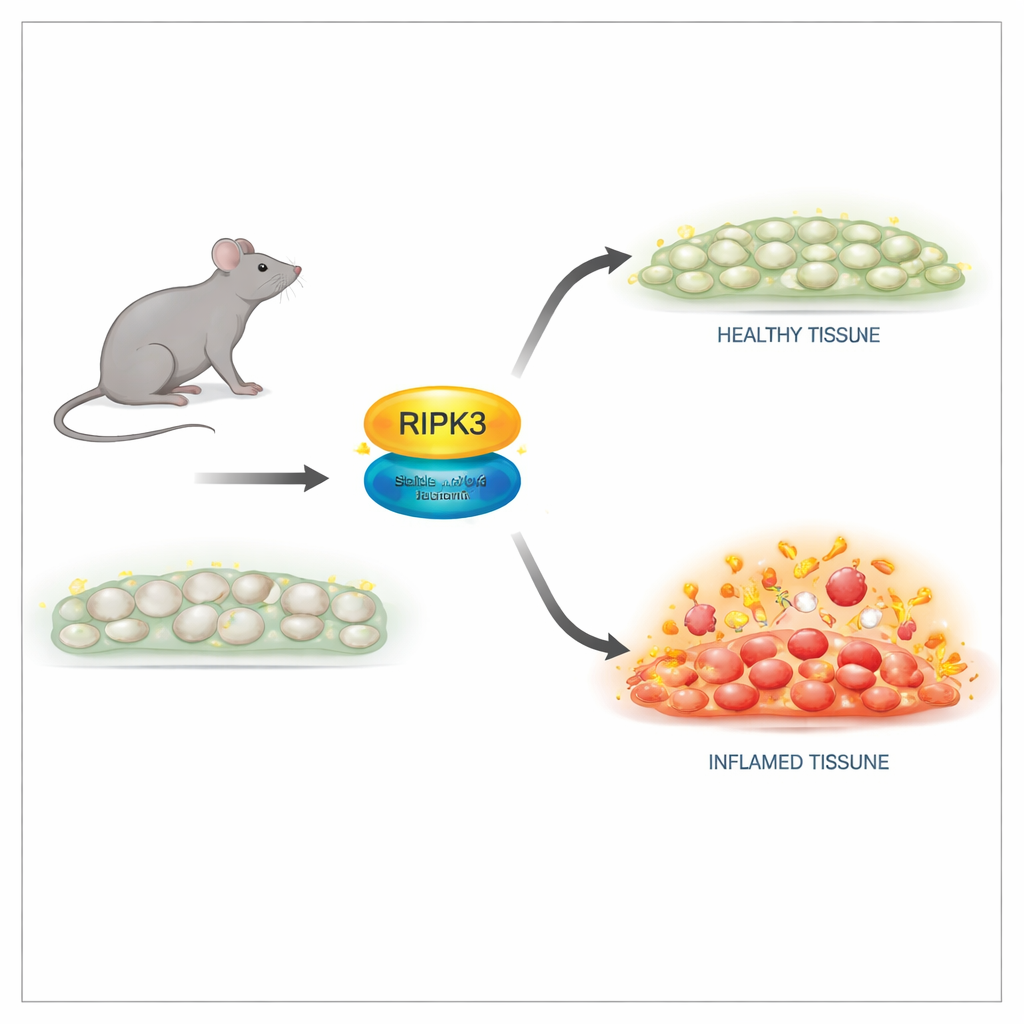
En säkrare metod för att stänga av den dödande funktionen
Forskarna konstruerade möss med en subtil förändring i RIPK3-proteinet vid en enda position, kallad D143N, som slår ut dess enzymaktivitet samtidigt som strukturen bevaras. I celler från dessa möss var RIPK3-nivåer och vävnadsarkitektur normala, och djuren föddes och växte som sina friska kullsyskon. Viktigt nog var celler med D143N-versionen helt resistenta mot flera necroptos-utlösare, inklusive signaler från tumörnekrosfaktor (TNF), toll-liknande receptorer och viral infektion. Den mutanta RIPK3 kunde inte längre aktivera sin nedströms partner eller bilda det destruktiva komplex som krävs för membranruptur, men den framkallade inte spontan apoptos, och undvek därmed de dödliga bieffekter som setts i äldre RIPK3-mutander.
Att skilja utveckling från sjukdom
En av RIPK3:s mest kända roller är i embryon som saknar ett annat viktigt protein, kaspas-8: utan kaspas-8 dödar RIPK3-driven necroptos embryot. I denna studie räddade införandet av D143N-versionen av RIPK3 helt dessa annars icke livsdugliga möss. De utvecklades normalt och var fertila, vilket visar att RIPK3:s dödande aktivitet är dispensabel för normal utveckling när dess struktur bevaras. Men när vuxna möss utsattes för högdos TNF för att inducera ett chockliknande inflammatoriskt syndrom ändrades bilden. Djur som helt saknade RIPK3 var starkt skyddade mot död, vävnadsskada och inflammatoriska mediatorer i blodet. Möss med D143N-versionen, trots frånvaro av necroptos, var bara delvis skyddade. Det indikerade att RIPK3:s icke-dödande, scaffold-roll fortfarande bidrog till att driva inflammation.
Scaffold-signalering som eldar på inflammationen
För att förstå detta icke-dödliga bidrag granskade teamet genaktiviteten i tarmen hos TNF-behandlade möss. I RIPK3-deficienta djur dämpades många inflammatoriska gener kraftigt. I D143N-möss var dock dämpningen svagare, och gener kopplade till interferon- och medfödda immunsvar förblev mer aktiva. På proteinnivå aktiverade TNF starkt JAK–STAT1- och ERK-signalvägarna i både normala och D143N-möss, men denna aktivering saknades nästan helt när RIPK3 raderades helt. Detta visade att även utan sin dödande funktion hjälper RIPK3:s fysiska närvaro i signaleringskomplex att föra TNF-signaler vidare till ett proinflammatoriskt program via JAK–STAT1.
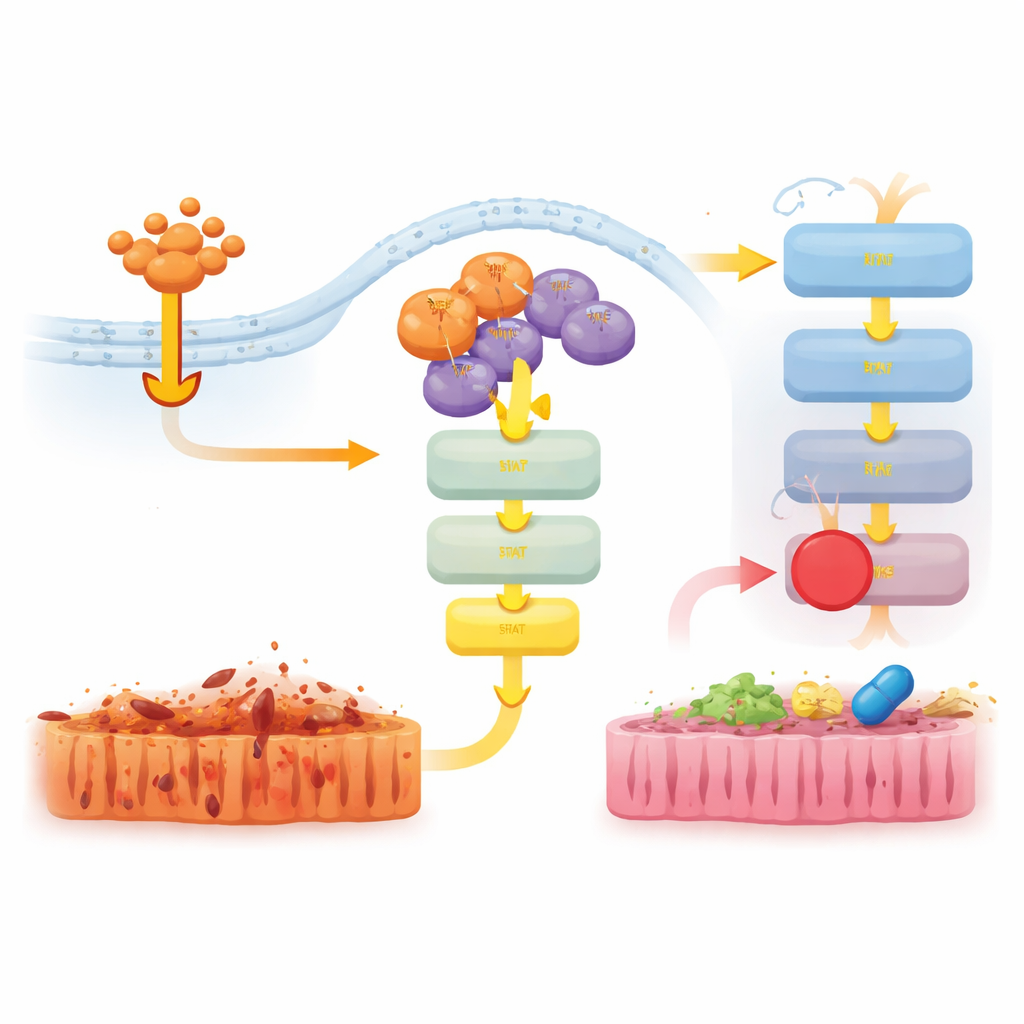
Att dämpa skadliga signaler med riktade läkemedel
Forskarna undersökte sedan om blockering av dessa nedströms vägar kunde mildra sjukdomen hos D143N-möss som genomgick TNF-inducerad chock. Behandling med en JAK1/2-hämmare, men inte en ERK-hämmare, minskade kroppstemperaturfall, sänkte nivåerna av den inflammatoriska molekylen IL-6 och minskade tarmskador och celldöd. En separat hämmare riktad mot ett annat protein, RIPK1, skyddade också starkt mössen och dämpade JAK–STAT1- och ERK-aktivering. Tillsammans tyder dessa resultat på att RIPK3:s scaffold-funktion samarbetar med RIPK1 för att aktivera JAK–STAT1 och driva inflammation, och att avbrott i denna signalering kan minska vävnadsskada även när necroptos redan är blockerad.
Vad detta betyder för framtida behandlingar
I åratal har RIPK3 främst betraktats som en omkopplare för en toxisk form av celldöd, och läkemedelsutveckling har fokuserat på att stänga av dess enzymaktivitet. Denna studie visar att det kanske inte räcker: RIPK3 kan fortfarande fungera som en fysisk plattform som förstärker inflammatoriska signaler genom JAK–STAT1 och bidra till chock och vävnadsskada. Den nya D143N-musmodellen blottlägger dessa dubbla roller med ovanlig tydlighet och etablerar ett kraftfullt verktyg för att studera när och hur varje funktion spelar roll i olika sjukdomar. För patienter antyder arbetet att kombinationer av läkemedel som riktar sig mot RIPK3 eller RIPK1 ihop med blockerare av JAK–STAT1 kan dämpa skadlig inflammation mer effektivt i tillstånd drivna av TNF och närliggande cytokiner.
Citering: Du, Y., Li, J., Zhao, C. et al. A viable kinase-inactive RIPK3 D143N mouse model reveals its scaffold function in driving TNF-induced inflammatory disorder. Cell Death Discov. 12, 107 (2026). https://doi.org/10.1038/s41420-026-02962-x
Nyckelord: RIPK3, necroptos, inflammation, TNF-chock, JAK-STAT1